Case Report
Focal Ab-amyloid deposition precedes cerebral microbleeds and Superficial siderosis: a case report

Parnesh Raniga1, Patricia Desmond2, Paul Yates3, Olivier Salvado1, Pierrick Bourgeat1, Jurgen Fripp1, Svetlana Pejoska3, Michael Woodward4, Colin L Masters5, Christopher C Rowe3,4,6 and Victor L Villemagne3,5,6*
1CSIRO Preventative Health National Research Flagship, Australian e-Health Research Centre-BioMedIA, Brisbane, QLD, Australia
2Department of Radiology, University of Melbourne and the Royal Melbourne Hospital, Melbourne, VIC, Australia
3Department of Molecular Imaging & Therapy, Austin Health, Melbourne, VIC, Australia
4Department of Aged Care, Austin Health, Melbourne, VIC, Australia
5The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, VIC, Australia
6Department of Medicine, Austin Health, University of Melbourne, VIC, Australia
*Address for Correspondence: Victor L Villemagne, Department of Molecular Imaging & Therapy, Austin Health, 145 Studley Road, Heidelberg, Vic. 3084, Australia. Tel: +61-3-9496 3321; Fax: +61-3-9458 5663; Email: [email protected]
Dates: Submitted: 18 September 2017; Approved: 12 October 2017; Published: 13 October 2017
How to cite this article: Raniga P, Desmond P, Yates P, Salvado O, Bourgeat P. et al. Focal Ab-amyloid deposition precedes cerebral microbleeds and Superficial siderosis: a case report. J Neurosci Neurol Disord. 2017; 1: 039-044. DOI: 10.29328/journal.jnnd.1001007
Copyright License: © 2017 Raniga P, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Brain imaging; Alzheimer’s disease; Ab-amyloid; PiB; cerebral amyloid angiopathy
Abstract
This case report presents in-vivo findings on the spatial and temporal relationship between focal Ab-amyloid deposition, cerebral micro-haemorrhages and superficial siderosis. A 65-year-old woman underwent 11C-PiB PET scans that revealed an atypical focal and asymmetrical pattern of Ab-amyloid deposition and MRI scans that revealed cerebral micro-haemorrhages and superficial siderosis. Almost all micro-haemorrhages were associated with focal Ab-amyloid deposition. Follow-up 11C-PiB PET and MRI scans showed progression of the disease. We speculate that Abamyloid deposition affects the structural integrity of arterioles, thereby predisposing them to micro haemorrhages. In support of this hypothesis, progression of MRI lesions was observed only in areas associated with Ab-amyloid deposition.
Introduction
Cerebral amyloid angiopathy (CAA), a condition where Ab-amyloid deposits in and around the media of small arteries and arterioles of the cerebral cortex and leptomeninges has been found to be present in most patients with Alzheimer’s disease (AD) [1]. To this date, neuropathologic examination of the brain remains the only definitive method for diagnostic confirmation of CAA. However the combination of Ab-amyloid imaging with 11C-PiB-PET [2] and T2* susceptibility weighted (SWI) MR imaging [3], allows for the concomitant assessment of molecular and structural changes in-vivo.
The combination of cerebral micro-haemorrhages (MH)-small focal areas of blood extravasation-and superficial siderosis (SS), characterised by hemosiderin deposits in the subpial layers of the brain and appearing as a hypo-intense rim on the surface of the brain on MRI, have been suggested as radiological markers for CAA [4]. Indeed SS has been shown to be prevalent in subjects with CAA, and is rare in non-CAA forms of intracerebral haemorrhage [5]. Moreover, MH and SS have been shown to co-localize with high PiB retention [6].
There have not been any reports in the literature of longitudinal follow-up in subjects evaluating the relationship between MH, SS and Aβ-amyloid deposition. In this case-study we report the spatial and temporal relationship between Aβ-amyloid deposition and radiological markers of MH and SS.
Matherial and Methods
Clinical evaluation
Studies were approved by the Austin Health Human Research Ethics Committee. Written informed consent was obtained prior to the examinations. Medical history was obtained from the participant, carer and from physical examination. Vascular risk factors were identified using self-report, physical examination and laboratory findings: hypertension, hypercholesterolemia, diabetes, current smoking history, atrial fibrillation, prior or current history of vascular disease (coronary or peripheral vascular disease), and dichotomised as present or absent according to published diagnostic guidelines.
Neuropsychological evaluation and Imaging data
Details of neuropsychological evaluation and imaging data are provided in the supplementary materials. Briefly, a neuropsychological battery of assessments was conducted including MiniMental State Examination (MMSE) and Clinical Dementia Rating (CDR) as previously described [7]. 18F-FDG and 11C-PiB PET scans were obtained on a Phillips Allegro™ PET camera as previously described [2,8]. MRI imaging was obtained on a 3Tesla Siemens scanner with sequences including a 3D T1-Weighted Magnetization Prepared Rapid Gradient Echo (MPRAGE), a SWI and a Fluid Attenuated Inversion Recovery (FLAIR) scan.
Genetic profile
ApoE allele subtype was determined by PCR amplification of genomic DNA.
Image analysis
All MRI were inspected blind to clinical and 11C-PiB scan findings. Number and location of MH and SS were recorded for each SWI scan by an expert neuroradiologist (P.D). A difference map between the 2009 and 2007 SWI scans was computed and manually thresholded. The difference map and the manual segmentation were used to highlight evolving (change>5%), stable (change<5%) and new MH and SS. WMH volumes were computed from manually segmented FLAIR images.
Results
Case report
A 65 -year-old female with family history of AD, presented with memory complaints since the beginning of 2005. Physical examination in August 2005 revealed no focal neurological signs, normal blood tests (including B12, folate and thyroid function), normal blood pressure (140/85 mm Hg) and no cardiovascular risk factors. Participant reported no use of anticoagulant medication. Her MMSE at that stage was 28/30, CDR was 0.5 and she was classified as suffering mild cognitive impairment (MCI). She underwent an 18F-FDG PET study that was suggestive of AD, with evidence of mild to moderate temporoparietal hypo metabolism, with sparing of the frontal, sensorimotor and occipital cortices (Figure 1A). Seven months after initial presentation, she underwent her first PiB study while her MMSE decreased to 27/30. There was progressive cognitive decline and 18 months after initial presentation she was diagnosed as probable AD and started on Galantamine (dose increased up to 16 mg/d). She was subsequently enrolled in the AIBL study [9] and she received her second PiB PET scan and first set of MRIs at 24 months after initial presentation. At that stage, her MMSE was 22/30 with a CDR of 1.0; there were still no focal neurological signs and blood tests were normal. In April 2009, after her second set of MRIs and before her next 11C-PiB or neuropsychological evaluation, she was withdrawn from the study by her husband. The demographic information of the subject along with memory scores and quantitative imaging measures at different time-points are summarized in (Table 1).
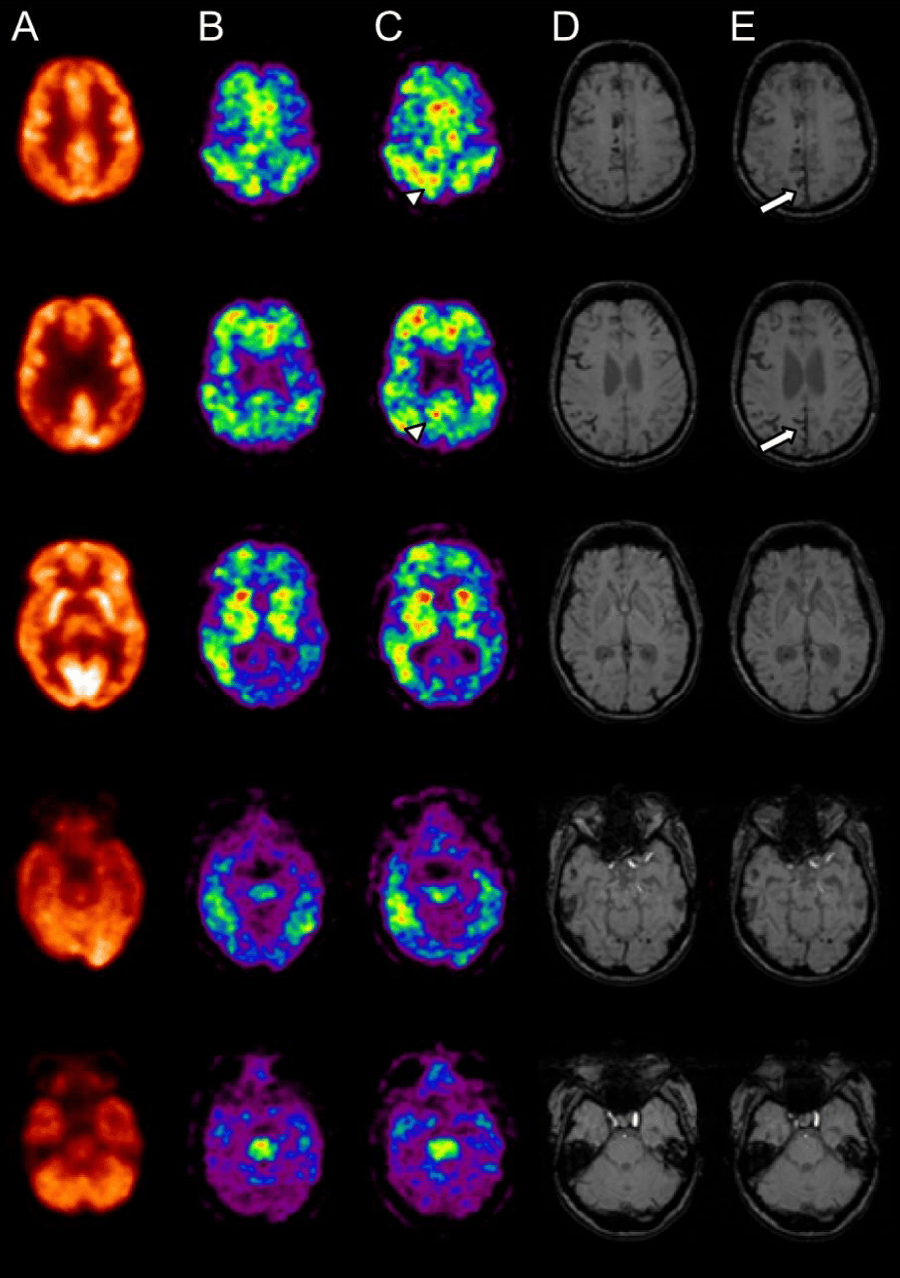
Figure 1: Representative PET-MRI coregistered images showing extent of glucose hypometabolism (a: Sept 2005); focal Ab deposition as measured by PiB (b: March 2006 and c: July 2007) and extension of MH and SS as measured by MRI SWI (d: August 2007 and e: April 2009). Follow up MRI SWI (April 2009 (e)) shows new MH (white arrows) in those regions shown to have focal Ab deposition (PiB PET July 2007(c)) (arrow heads).
| Table 1: Demographic and imaging information at the different time-points of the study. | ||||
| Gender | Female | |||
| ApoE | e3/e4 | |||
| Study Times | ||||
| Sep-05 | Mar-06 | Aug-07 | Apr-09 | |
| Age | 65 | 66 | 67 | 69 |
| Dx | MCI | MCI | AD | AD |
| MMSE | 28 | 27 | 22 | 19 |
| CDR | 0.5 | 0.5 | 1.0 | |
| CDR SOB | 1.5 | 4.5 | ||
| Comp Ep Mem | -1.9 | -3.3 | ||
| Comp Non Mem | -1.7 | -1.8 | ||
| PiB SUVRpons | 0.9 | 1.0 | ||
| GM vol (ICV) | 507.0 mL (0.30) | 489.0 mL (0.29) | ||
| Hipp vol (ICV) | 6.1 mL (0.0041) | 5.2 mL (0.0035) | ||
| Ventricular vol (ICV) | 42.3 mL (0.034) | 57.0 mL (0.046) | ||
| WMH vol (ICV) | 43.3 mL 0.035) | 50.2 mL (0.041) | ||
| MMSE=Mini Mental State Exam; CDR=Clinical Dementia Rating; CDR-SOB=Clinical Dementia Rating-Sum of Boxes; Comp Ep Mem=Composite Episodic Memory score; Comp Non Mem=Composite Non-memory score; GM=Total gray matter Hipp=Hippocampus; WMH=White matter hyperintensities | ||||
Imaging studies
PiB. Visually, and in clear contrast to the diffuse distribution observed in age-matched AD patients, the PiB PET study from 2006 revealed multiple foci of cortical 11C-PiB retention, in frontal, striatum, anterior cingulate, temporal and parietal cortices, particularly on the right side, with sparing of the sensorimotor and occipital cortices (Figure 1B). The PiB PET study from 2007 showed similar but more extensive focal 11C-PiB retention (Figure 1C). This was also evidenced through Zscore maps of the subject’s follow-up PIB scan compared to AD patients (Supplementary Figure 1).
The 11C-PiB scan from 2006 was co-registered to its 2007 follow-up scan. The 2007 11C-PiB scan was threshold to SUVRpons of 0.85 which was 20% higher than the optimal neocortical SUVRpons threshold of 0.71 used to separate healthy control subjects with high PiB retention from subjects with low PiB retention computed using hierarchical cluster analysis. (See Supplementary Materials) The mask of the thresholded region from the 2007 scan was used to sample both the co-registered 2006 and the 2007 scans. The average SUVRpons within the mask was 0.85 for the 2006 scan and 0.95 for the 2007 scan (an overall increase of 11.7%).
MRI. In 2007, the T1-weighted MRI showed mild cortical, as well as left hippocampal and left entorhinal cortex atrophy suggestive of early AD. On FLAIR, there was extensive white matter lesions with white matter hyper intensities (WMH) quantified as 43.3 ml; and no evidence of ischemic infarcts. On SWI, there were multiple MH in a distribution suggestive of CAA and extensive SS (Figure 1D) (See supplementary materials for topography of MH). Ventricular volume was measured at 42.3 ml.
Compared with the MRI from 2007, in 2009, there was increased sulcal dilatation and ventricular enlargement (57.0 ml), and progressive hippocampal atrophy bilaterally. There was also progression in the white matter changes in the corona radiata bilaterally (WMHV=50.2 ml) and six new MH were identified on the SWI image in the left temporal, lateral occipital, right occipital, left frontal, right frontal and right parietal lobes. Two new sites of SS in the posterior parietal lobe were also observed. Two lacunar infarcts were evident with one overlapping a large MH suggesting a local haemorrhagic change.
The overlap between the 11C-PiB scan from 2007 and MRI SWI scans from 2007 are shown in (Supplementary Figure 1). As can be observed, SWI lesions, especially superficial siderosis, overlap the thresholded PiB retention prominently. Of note is a large area of increased PiB signal in the posterior cingulate-posterior parietal region. Although this area has no SWI lesions on the initial scan, on the follow-up MRI it is associated with a large area of superficial siderosis. Furthermore, there are several new MH, all in regions associated with Ab-amyloid deposition on the 2007 PiB scan. When comparing changes in SWI lesions with changes in PiB retention, those SWI lesions that progressed in 2009 also showed a higher increase in focal PiB retention (Supplementary Table 1).
Discussion
As previously reported MH and SS were intimately associated with Ab-amyloid deposition [6,10]. While lobar MH are a frequent finding in AD patients or even in cognitively-normal older individuals, they are strongly associated with increasing age and Ab-amyloid deposition [11]. This association between Ab-amyloid and vascular lesions has crucial implications not only for the selection and risk stratification of individuals undergoing anticoagulant therapies, but also in those enrolled in anti-Ab-amyloid therapeutic trials [12]. It is thought that the typical Ab-amyloid deposits in and around the media of small arteries and arterioles of the brain weakens the vessel walls predisposing to MH.
The focal and asymmetrical nature of the Ab-amyloid deposition in this case is of particular interest, and in contrast to the diffuse cortical pattern usually observed in AD. Conversely, the clinical history was consistent with Alzheimer’s type dementia, and without classical features of stepwise cognitive decline or progressive neurological signs to suggest a vascular aetiology. While it has been proposed that the regional pattern of Ab-amyloid deposition may separate AD from CAA, without histopathological confirmation it is impossible to ascertain if the 11C-PiB retention in our patient is mainly attributable to vascular deposits, plaques, or both.
Much more striking are the observations derived from the longitudinal aspect of the study, where areas with focal Ab-amyloid deposition and no MH in the respective PiB and MRI SWI studies in 2007, matched the new MH and larger areas of SS observed in the MRI SWI study from 2009. All this imaging data points to CAA, both by the location of the MH-areas largely towards the surface of the cerebral cortex where the interstitial fluid normally diffuses on its way out of the brain- as well as for the SS usually the end result of repeated small haemorrhages, most likely to happen in leptomeningeal arteries as they transverse from the subarachnoid space penetrating into the parenchyma and forming cortical arteries. Both the leptomeningeal and cortical arteries have been found to be heavily involved in CAA [12].
The distribution of WMH in this patient is in agreement with previous reports on subjects with AD, CAA and vascular dementia [18]. There were no WMH in the brainstem on either the 2007 or 2009 MRI scans; an observation that has been linked with increased likelihood of cerebrovascular disease [14]. The involvement of capillaries in CAA, more prominent in subjects with APOE e4 alleles, has been linked with the development of occlusions [15] and presumably ischemia related white matter hyperintensities.
An increase of almost 12% in Ab-amyloid deposition was also detected in the 16 months between the 2006 and 2007 PiB studies, higher than our reported 5.7% in a large cohort of AD patients [16] but similar to the increase in Ab-amyloid deposition reported by Rinne and colleagues in AD patients on the placebo arm of their therapeutic trial [17]. This increase in neocortical Ab-amyloid deposition was accompanied by a decline in cognitive performance leading to the clinical diagnosis of AD.
This study demonstrates that the combination of different imaging techniques provides essential complementary information crucial to ascertain the underlying pathological disorder [18]. Further longitudinal population-based studies combining clinical information with these neuroimaging techniques will help better elucidate the relationship between Ab-amyloid deposition and microhemorrhages and superficial siderosis.
Acknowledgements
Funding: PET studies were supported in part by funds from the Austin Hospital Medical Research Foundation, Neurosciences Victoria, the University of Melbourne, and the AIBL Study (http://www.aibl.csiro.au/).
Ethics approval: PET imaging studies were approved by the Austin Health Human Research Ethics Committee. Written informed consent was obtained from the appropriate next of kin prior to the scans.
References
- Jellinger KA, Attems J. Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J Neurol Sci. 2005; 229-230: 37-41. Ref.: https://goo.gl/ro46Px
- Smith DC, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007; 68: 1718-1725. Ref.: https://goo.gl/XYWWFB
- Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the Boston Criteria. Neurology. 2001; 56: 537-539. Ref.: https://goo.gl/7VzhiH
- Feldman HH, Maia LF, Mackenzie IRA, Forster BB, Martzke J, et al. Superficial Siderosis: A Potential Diagnostic Marker of Cerebral Amyloid Angiopathy in Alzheimer Disease. Stroke. 2008; 39: 2894-2897. Ref.: https://goo.gl/YzysMy
- Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010; 74: 1346-1350. Ref.: https://goo.gl/xgoRSu
- Dhollander I, Nelissen N, Van Laere K, Peeters D, Demaerel P, et al. In vivo amyloid imaging in cortical superficial siderosis. J Neurol Neurosurg Psychiatr. 2011; 82: 469-471. Ref.: https://goo.gl/taca5n
- Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, et al. β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007; 130: 2837-2844. Ref.: https://goo.gl/n7uZ61
- Ng S, Villemagne VL, Berlangieri S, Lee S-T, Cherk M, et al. Visual Assessment Versus Quantitative Assessment of 11C-PIB PET and 18F-FDG PET for Detection of Alzheimer’s Disease. J Nucl Med. 2007; 48: 547-552. Ref.: https://goo.gl/Chr5pR
- Ellis KA, Bush AI, Darby D, De Fazio D, Foster J, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr. 2009; 21: 672-687. Ref.: https://goo.gl/QGkr7F
- Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RNK, et al. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol. 2010; 68: 545-548. Ref.: https://goo.gl/k5XJau
- Yates PA, Sirisriro R, Villemagne VL, Farquharson S, Masters CL, et al. For the AIBL Research Group Cerebral microhemorrhage and brain β-amyloid in aging and Alzheimer disease. Neurology. 2011; 77: 48 -54. Ref.: https://goo.gl/GNpjEa
- Weller RO, Preston SD, Subash M, Carare RO. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res Ther. 2009; 1: 6. Ref.: https://goo.gl/BG4MFa
- Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, et al. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke 2008; 39: 1127-1133. Ref.: https://goo.gl/wFnkzm
- Smith EE. Leukoaraiosis and Stroke. Stroke. 2010; 41: S139-143. Ref.: https://goo.gl/BC7A9C
- Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, et al. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging. 2009; 30: 1936-1948. Ref.: https://goo.gl/7PM88E
- Villemagne VL, Pike KE, Chételat G, Ellis KA, Mulligan RS, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. 2011; 69: 181-192. Ref.: https://goo.gl/rLdZf1
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, et al. 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010; 9: 363-372. Ref.: https://goo.gl/gEJoWh
- Jack CR, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008; 131: 665-680. Ref.: https://goo.gl/LZQGez