
More Information
Submitted: August 23, 2021 | Approved: September 07, 2021 | Published: September 08, 2021
How to cite this article: Dave S, Zhou A. Differential roles of trithorax protein MLL-1 in regulating neuronal Ion channels. J Neurosci Neurol Disord. 2021; 5: 089-093.
DOI: 10.29328/journal.jnnd.1001057
Copyright License: © 2021 Dave S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: MLL-1; Epigenetics; Polycomb group proteins; Trithorax group proteins; Repressor; Potassium channel; Ischemic stress

Differential roles of trithorax protein MLL-1 in regulating neuronal Ion channels
Sonya Dave1,2 and An Zhou1*
1Department of Neurobiology, Morehouse School of Medicine, Atlanta, Georgia, USA
2Department of Biology, Emory University, Atlanta, Georgia, USA
*Address for Correspondence: An Zhou, Ph.D, Department of Neurobiology, Neuroscience Institute, Morehouse School of Medicine, 720 Westview Drive SW, Atlanta, GA, USA, Tel: (404)7565722: Fax: (404)7521041; Email: [email protected]
Repressive regulation of potassium channel genes by Polycomb group (PcG) proteins contributes to PcG protein-mediated neuroprotection against neuronal ischemic injury, as seen in an ischemic stroke. Here we asked the question whether Trithorax group (TrxG) proteins, the antagonistic partners of PcG proteins (i.e, epigenetic activators targeting the same genes) may also regulate potassium channels. Results of patch-clamp studies on cultured neuronal cells showed that inhibition of TrxG protein MLL-1 led to an increase in potassium channel activity, an unexpected effect for a presumed gene activator. In contrast, decreased sodium currents were observed with MLL-1 inhibition. Increased or decreased levels of potassium channel protein Kv2.1 or sodium channel protein Nav1.2, respectively, were seen with MLL-1 inhibition, as determined by immunocytochemistry. These results, for the first time, demonstrate an involvement of TrxG protein MLL-1 in regulating neuronal ion channels, potentially repressing potassium channel genes.
Ion channels play pivotal roles in regulating neuronal response to stresses, such as ischemic stress in brain ischemia. In the phenomenon of the so-called neuronal ischemic tolerance (modeled resistance against ischemic injury), a decrease in whole cell potassium (K+) currents and levels of a number of K+ channel genes or proteins has been observed in cultured cortical neurons in vitro and in mouse brains in vivo [1-3]. The possible involvement of multiple K+ channel genes in producing ischemic tolerance suggests an epigenetic mechanism underlying the observed changes in K+ channels. In a previous study on effectors of ischemic tolerance, we have determined neuroprotective roles of several polycomb group (PcG) proteins (namely BMI-1, RING1B and SCMH1), the known epigenetic repressors [2]. Our recent results show that EZH2, another PcG protein, is also neuroprotective (unpublished results described in a separate manuscript). For each of the afore-mentioned PcG proteins, over-expressing or silencing results in a decrease or increase, respectively, in K+ currents in cultured neuronal cells [2]. These findings have signified important roles of PcG protein-mediated, epigenetic regulation of neuronal ion channels. Such regulation in turn may determine how neuronal cells respond to harmful ischemic stresses as seen in an ischemic stroke.
PcG proteins potentially target more than a thousand genes, including many K+ channel genes [4]. They exert their repressive roles by concerted modifications of specific histone proteins in a residue-specific manner. Two well-studied, PcG protein-mediated histone modifications include mono-ubiquitination of histone H2A at Lysine 119 (H2K119mUb) by RING1B or BMI-1, and tri-methylation of H3 at Lysine 27 (H3K27me3) by EZH2; both H2K119mUb and H3K27me3 are repressive epigenetic marks. Interestingly, for genes that are repressively regulated by PcG proteins, they are also subject to activating regulation by another group of epigenetic regulatory proteins, the trithorax group (TrxG) proteins [5-9]. Like PcG proteins, TrxG proteins also modify histone proteins as a mean to installing activating epigenetic marks; examples include H3 tri-methylation at Lysine 4 (H3K4me3) by MLL-1 and at Lysine 36 (H3K36me3) by ASH1L, respectively. Our early, limited data suggest that, in ischemic-injured or ischemic-tolerant brains, the levels of PcG and TrxG proteins change in the opposite direction [10]. These observations have led us to ask the question whether TrxG proteins may regulate neuronal ion channels in effects opposing to that of PcG proteins, and subsequently the neuronal response to ischemia.
To investigate this question, it is essential that the effects of TrxG proteins be examined at functional levels. In this work, we took advantage of a commercially available pharmacological inhibitor for TrxG protein MLL-1, a H3K4 methyltransferase, and tested the effects of MLL-1 inhibition on electrophysiological properties of differentiated neuroblastoma NS20Y cells. Differentiated NS20Y cells have many characteristics that are comparable to those of primary cortical neurons, such as response to ischemic stress and biosynthesis of neuropeptides [2,11-13]. Previously, we have used this cell line to establish repressive regulation of K+ channels by PcG proteins. The current results show that inhibition of MLL-1 produced, unexpectedly, an increase in K+ currents, challenging MLL-1 as a default gene activator being a TrxG protein. The results also suggest the potential significance of investigating MLL-1’s roles in brain ischemia.
Cell cultures
Mouse brain-derived neuroblastoma NS20Y cells (Sigma, St. Louis, MO) were maintained under normoxic conditions in DMEM media supplemented with 10% fetal bovine serum. Differentiation was achieved by the addition of 0.5 mM 8-CPT-cAMP (Abcam, Cambridge, MA) to the culture, as previously described [2]. For MLL-1 inhibition, differentiated cells were incubated with 100 µM MM-102 (HMTase Inhibitor IX MM-102, Calbiochem, San Diego, CA; dissolved in DMSO and diluted with culture media) overnight (approximately 16 hours). Control cells were incubated with culture media containing 0.1% DMSO.
Immunocytochemistry (ICC)
The effectiveness of MLL-1 inhibition on specific histone modification (H3K4me3) and cellular levels of selected ion channel proteins were examined by ICC analysis following standard protocols as previously described [2]. Briefly, cells were fixed with 4% polyformaldehyde in PBS, permeabilized, and analyzed with specific antibodies as follows (antibody against-, dilution, vendor, catalog no): H3K4me3, 1:400, Abcam, Ab6000; Kv2.1, 1:200, NeuroMab, Clone 89/34; Nav1.2, 1:400, Origene, TA322242. For immunofluorescent detection, Dylight 594-conjugated secondary antibodies were used (1:800, ThermoScientific, 35510 and 35560 for anti-mouse IgG and anti-rabbit IgG, respectively). After incubation with appropriate antibodies, the specimens were mounted with a DAPI-containing solution to reveal nuclei, and examined under a Zeiss Axioskop2 fluorescence microscope.
Electrophysiology
Patch-clamp recordings were conducted on MM-102-treated or control cells that were plated on the same day. For Na+ channel recording, the extracellular solution (pH 7.37~7.43, 320~330 mOsm) contained (in mM): NaCl (140), KCl (5.4), CaCl2 (2), MgCl2 (1), HEPEPS (20), Glucose (10), in the presence of calcium (Ca++) channel blocker CdCl2 (0.1). For K+ channel recording, the extracellular solution included an addition of 1 µM Tetrodotoxin, a Na+ channel blocker and 100 µM CdCl2. The intracellular solutions (pH 7.27~7.33, 290~300 mOsm) contained (in mM): EGTA (0.5), HEPES (10), NaCl (15), MgATP (2), Na2GTP (0.3), either with K+ channel blockers CsCl (120) and Tetra-ethylammonium-Cl (2) for Na+ recordings, or with K-Gluconate (120) for K+ channel recordings. MM-102 was not included in the extracellular solution during recording. Recordings were performed on a Digidata 1322 Digitizer and Axon Axopatch 200B amplifier (Molecular Devices, San Jose, CA) using 40% for both prediction and compensation. Leak subtraction was used for Na+ channel recordings.
Statistical analysis
Data were excluded from analysis if they met the following criteria: cells exhibited a resting membrane potential (RMP) equal or greater than -20 mV in K+ channel recordings, or were an outlier (using 2 times of standard deviation of the mean as a cutoff) in 2 or more of the following three parameters: RMP, I-Density, or Capacitance. I-density vs. voltage traces were analyzed with two-way ANOVA with Bonferroni post-hoc analysis, with the assistance of Prism 6.0 software (GraphPad, San Diego, CA); p ≤ 0.05 is defined as significant.
First, in differentiated NS20Y cells, the effectiveness of MLL-1 inhibition by MM-102 was verified by examining cellular levels of H3K4me3, whose production relies on the activity of MLL-1. As demonstrated in Figure 1a, MM-102-treated cells showed decreased levels of H3K4me3 proteins.
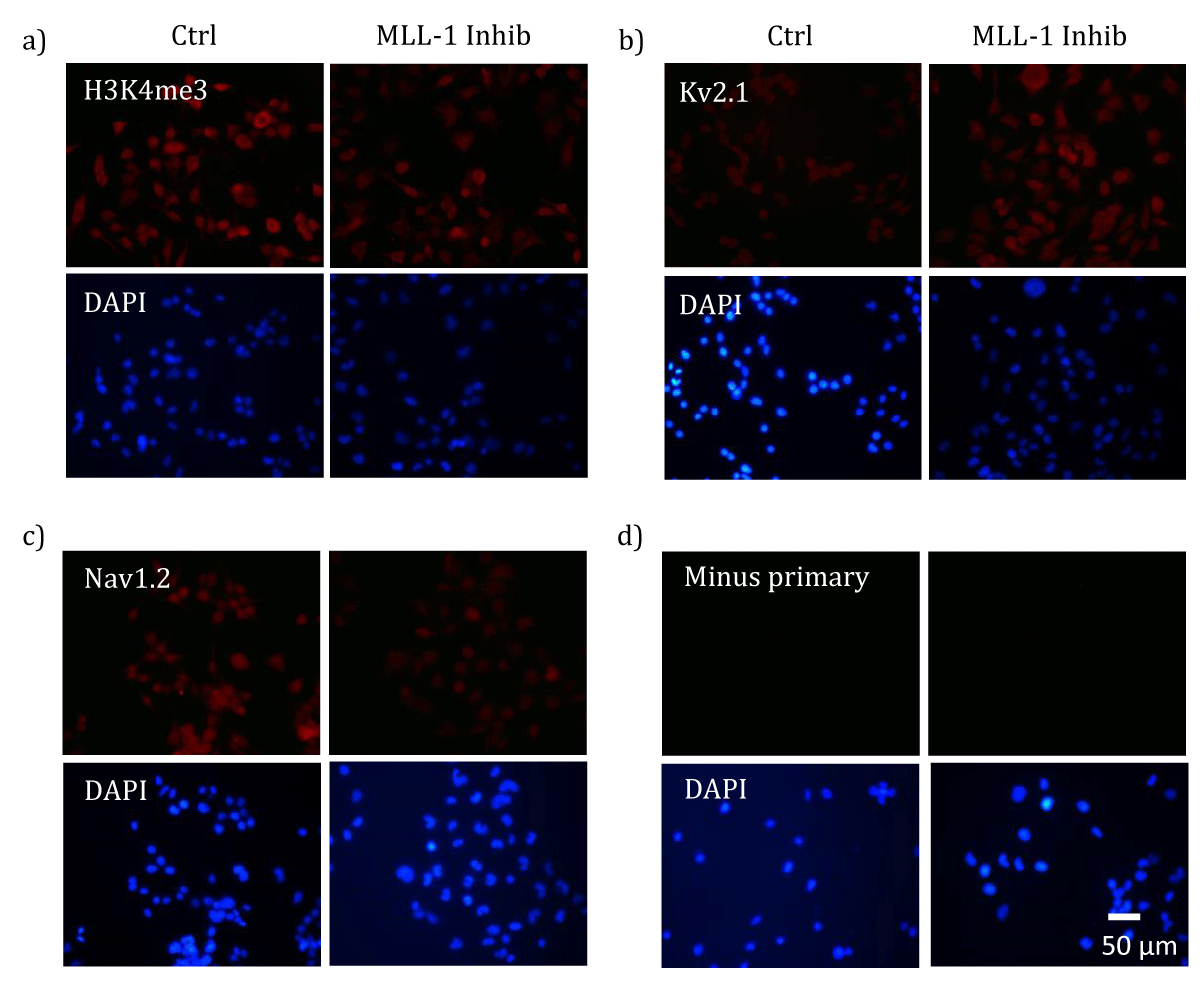
Figure 1: MLL-1 inhibition-induced changes in cellular levels of H3K4me3 and ion channel proteins. Differentiated NS20Y cells were treated with either 100 µM MM-102 (Inhib) or vehicle (0.1% DMSO, Ctrl). ICC analysis of H3K4me3 (a) or Kv2.1 (b) or Nav1.2 (c) was performed using appropriate primary antibodies and Dylight 594-conjugated secondary antibodies (a and b, anti-mouse; c, anti-rabbit). A negative control (d, Minus primary) was included by incubating the specimen with secondary antibody alone in absence of the primary antibody. After incubation with antibodies, the specimens were mounted with a DAPI-containing solution to reveal nuclei, and examined under a fluorescence microscope. Similar results were obtained in 2-3 independent cultures. Scale bar represents 50 µm.
MLL-1 inhibition resulted in increased K+ currents and cellular levels of Kv2.1 proteins
When currents of voltage-gated K+ channels (Kv) were analyzed with voltage clamp recording, a significant increase was observed in MM-102-treated cells when compared with that in control cells (Figure 2a). An increase in the levels of Kv2.1 proteins was seen in MM-102-treated cells (Figure 1b). On the other hand, currents attributed to the inward rectifier K+ channel (KIR) function was not affected by MLL-1 inhibition (Figure 2b). Hence, MLL-1 appears to have a repressive effect on the expression of Kv channel gene(s) in NS20Y cells, as suggested by changes in both channel activity and levels of Kv2.1 proteins.
MLL-1 inhibition resulted in decreased Na+ currents and cellular levels of Nav1.2 proteins
To assess whether a potentially repressive role of MLL-1 is a general effect to voltage-gated ion channels, we analyzed Na+ currents and cellular levels of Nav1.2, a voltage-gated Na+ channel (Nav) protein. Figure 2c shows that there was a significant decrease in Na+ currents in MM-102-treated cells when compared with that in control cells. Nav1.2 is widely expressed in neuronal cell populations in the brain. In NS20Y cells, its levels show differential changes under different ischemic conditions [1], and it presumably contributes to the production of Na+ currents. The results of ICC analysis of Nav1.2 showed that there was an increase in its levels in MM-102-treated cells, when compared with that in control cells (Figure 1c). Thus, in contrast to its effects on Kv channels, MLL-1 exhibits an activating role in regulating Na+ channel gene(s) in NS20Y cells.
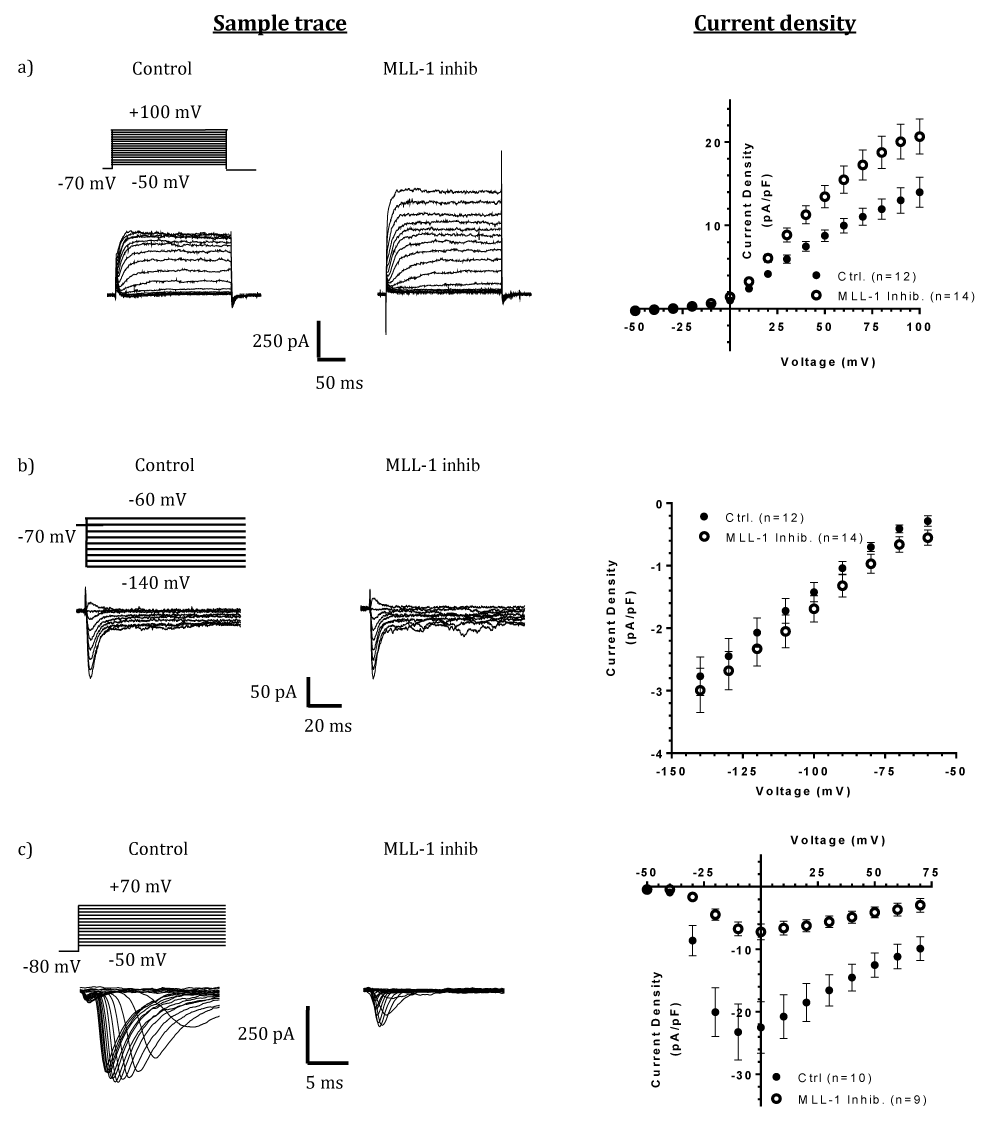
Figure 2: Effects of MLL-1 inhibition on K+ and Na+ currents in differentiated NS20Y cells. The left panels show sample traces for measurements of K+ (a), Na+ (b), and KIR (c) channels, respectively. The right panels demonstrate: (a) Steady-state K+ channel current density with voltage steps from -50 to +100 mV, which is indicative of Kv type of K+ channels. MLL-1 inhibition (open circles) significantly increased steady-state current density in main effect (F(1, 24) = 8.429, p = 0.0078) in a voltage dependent manner (interaction of MLL-1 inhibition X Voltage, F(15, 360) = 6.436, p < 0.0001), with significance from +50 to +100 mV, inclusive (+50 mV, p = 0.0272; +60 mV, p = 0.0036, +70 mV, p = 0.0004, +80 and +90 mV, p < 0.0001; +100 mV, p = 0.0001) when analyzed with two-way ANOVA. (b) Na+ channel current density (normalized with cell size) in response to voltage steps from -50 to +70 mV. MLL-1 inhibition (open circles) significantly decreased the peak current density in a voltage-dependent manner (main effect, F(1,17) = 15.37, p = 0.0011; interaction of MLL-1 inhibition X Voltage, F(18, 306) = 7.931, p < 0.0001; -20 to +10 mV, p < 0.0001; +20 mV, p = 0.0002; +30 mV, p = 0.0013; +40 mV, p = 0.0087; +50 mV, p = 0.0416). (c) Peak K+ channel current density with voltage steps from -60 to -140 mV, which is indicative of the KIR type K+ channel, and for which MLL-1 inhibition did not result in changes in current density (main effect, F(1, 24) = 0.9399, p = 0.3420; interaction of MLL-1 inhibition X Voltage, F(8,192) = 0.03859, p = 0.9999). Numbers in parentheses indicate the total numbers of cells analyzed; cells were collected from at least two independent cultures..
Unexpected, repressive effects of MLL-1 on K+ channel
It is interesting that, in differentiated NS20Y cells, the effects of MLL-1 inhibition differ between K+ and Na+ channels. The opposite changes in levels of Kv2.1 and Nav1.2 proteins (note that both are voltage-gated ion channel proteins), respectively, with MLL-1 inhibition implicate that MLL-1 may potentially act as a repressor for K+ channel gene(s) but an activator for Na+ channel gene(s). The increase in K+ currents with MLL-1 inhibition is in contrast to what one would expect, if MLL-1 is categorically an epigenetic activator being a TrxG protein. However, there have been studies in which MLL-1 shows similar roles to that of PcG proteins. In tumorgenesis, MLL-1 is known to play an oncogenic role, like PcG proteins typically do [14-18]. In brain tumor cells, MLL-1 enhances hypoxic responses, and silencing MLL-1 results in a decrease in hypoxia inducible factor (HIF) [16], known effects of PcG proteins. It should be noted that above-mentioned studies do not describe a direct effect, or the lack of it, of MLL-1 on gene expressions.
Mechanisms underlying MLL-1-mediated K+ channel repression
One may consider a number of possible underlying mechanisms in attempt to explain the observed repressive effects of MLL-1 on K+ channel. First, it is not known whether MLL-1 interacts directly with promoters of genes of interest or may regulate their expression through another regulatory protein. It is also possible that MLL-1 may activate a gene encoding a K+ channel inhibitory subunit. Second, as noted in Introduction, histone H3 carries both repressive and activating epigenetic marks, each of which may be gene-specific, and change in condition-specific, dynamic manners [9]. For a specific target gene, its expression often is determined by the balanced acts of multiple histone modifiers. The current results render the need of profiling epigenetic marks that occupy specific K+ or Na+ channel genes, using chromatin immunoprecipitation (ChIP) analysis combined with DNA sequencing or mass spectrometry analysis. Third, MLL-1 may be a noncanonical TrxG protein, that it may act like a PcG protein rather than as a TrxG protein, as there are examples for a PcG protein acting like a TrxG protein [19]. Also, the effects of PcG repressors and TrxG activators can potentially be context-dependent [20, 21]. Fourth, structurally, MLL-1 protein contains several regions that may potentially interact with (or recruit) gene repressor proteins including PcG protein BMI-1 and histone deacetylase 1 [22-25], whose actions may ablate MLL-1’s activity as a gene activator. While it remains to be shown whether such interactions may be more prevalent in the promoter regions of K+ channel genes as to that of Na+ channel genes, the possibility presents an interesting topic for future studies.
Summary
TrxG protein MLL-1 shows a regulatory role for both K+ and Na+ channels in neuronal cells, with a potentially repressive role for the former.
Authors acknowledge grant supports from American Heart Association (17GRNT33700277 to AZ) and National Institute of Health (NINDS U54NS060659 to Neuroscience Institute, MSM; NCMHD U54MD007602 to MSM). Authors thank Drs. Tiandong Leng and Zhigang Xiong for constructive discussions.
- Hernandez-Encarnacion L, Sharma P, Simon R, Zhou A. Condition-specific transcriptional regulation of neuronal ion channel genes in brain ischemia. Int J Physiol Pathophysiol Pharmacol. 2017; 9: 192-201. PubMed: https://pubmed.ncbi.nlm.nih.gov/29348796/
- Stapels M, Piper C, Yang T, Li M, Stowell C, et al. Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci Signal. 2010; 3: ra15. PubMed: https://pubmed.ncbi.nlm.nih.gov/20197544/
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003; 362: 1028-1037. PubMed: https://pubmed.ncbi.nlm.nih.gov/14522533/
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006; 20: 1123-1136. PubMed: https://pubmed.ncbi.nlm.nih.gov/16618801/
- Brand M, Nakka K, Zhu J, Dilworth FJ. Polycomb/Trithorax Antagonism: Cellular Memory in Stem Cell Fate and Function. Cell Stem Cell. 2019; 24: 518-533. PubMed: https://pubmed.ncbi.nlm.nih.gov/30951661/
- Geisler SJ, Paro R. Trithorax and Polycomb group-dependent regulation: a tale of opposing activities. Development. 2015; 142: 2876-2887. PubMed: https://pubmed.ncbi.nlm.nih.gov/26329598/
- Koch L. Gene regulation: Yin and Yang of Polycomb/Trithorax response elements. Nat Rev Genet. 2014; 15: 644-645. PubMed: https://pubmed.ncbi.nlm.nih.gov/25159600/
- Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 2010; 10: 669-682. PubMed: https://pubmed.ncbi.nlm.nih.gov/20865010/
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007; 128: 735-745. PubMed: https://pubmed.ncbi.nlm.nih.gov/17320510/
- Simon RP, Meller R, Zhou A, Henshall D. Can genes modify stroke outcome and by what mechanisms? Stroke. 2012; 43: 286-291. PubMed: https://pubmed.ncbi.nlm.nih.gov/22156698/
- O'Bryant Z, Leng T, Liu M, Inoue K, Vann KT, et al. Acid Sensing Ion Channels (ASICs) in NS20Y cells - potential role in neuronal differentiation. Mol Brain. 2016; 9: 68. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4920985/
- Sirianni MJ, Fujimoto KI, Nelson CS, Pellegrino MJ, Allen RG. Cyclic AMP analogs induce synthesis, processing, and secretion of prepro nociceptin/orphanin FQ-derived peptides by NS20Y neuroblastoma cells. DNA Cell Biol. 1999; 18: 51-58. PubMed: https://pubmed.ncbi.nlm.nih.gov/10025508/
- Zhou A, Minami M, Zhu X, Bae S, Minthorne J, et al. Altered biosynthesis of neuropeptide processing enzyme carboxypeptidase E after brain ischemia: molecular mechanism and implication. J Cereb Blood Flow Metab. 2004; 24: 612-622. PubMed: https://pubmed.ncbi.nlm.nih.gov/15181368/
- Ansari KI, Kasiri S, Mandal SS. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene. 2013; 32: 3359-3370. PubMed: https://pubmed.ncbi.nlm.nih.gov/22926525/
- Chan AKN, Chen CW. Rewiring the Epigenetic Networks in MLL-Rearranged Leukemias: Epigenetic Dysregulation and Pharmacological Interventions. Front Cell Dev Biol. 2019; 7: 81. PubMed: https://pubmed.ncbi.nlm.nih.gov/31157223/
- Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, et al. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012; 19: 428-439. PubMed: https://pubmed.ncbi.nlm.nih.gov/21836617/
- Papale M, Ferretti E, Battaglia G, Bellavia D, Mai A, et al. EZH2, HIF-1, and Their Inhibitors: An Overview on Pediatric Cancers. Front Pediatr. 2018; 6: 328. PubMed: https://pubmed.ncbi.nlm.nih.gov/30510924/
- Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015; 525: 206-211. PubMed: https://pubmed.ncbi.nlm.nih.gov/26331536/
- Malik B, Hemenway CS. CBX8, a component of the Polycomb PRC1 complex, modulates DOT1L-mediated gene expression through AF9/MLLT3. FEBS Lett. 2013; 587: 3038-3044. PubMed: https://pubmed.ncbi.nlm.nih.gov/23891621/
- Kingston RE, Tamkun JW. Transcriptional regulation by trithorax-group proteins. Cold Spring Harb Perspect Biol. 2014; 6: a019349. PubMed: https://pubmed.ncbi.nlm.nih.gov/25274705/
- Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell. 2017; 171: 34-57. PubMed: https://pubmed.ncbi.nlm.nih.gov/28938122/
- Cosgrove MS, Patel A. Mixed lineage leukemia: a structure-function perspective of the MLL1 protein, FEBS J. 2010; 277: 1832-1842. PubMed: https://pubmed.ncbi.nlm.nih.gov/20236310/
- McCarthy N. Leukaemia: MLL makes friends and influences. Nat Rev Cancer. 2010; 10: 529. PubMed: https://pubmed.ncbi.nlm.nih.gov/20677349/
- Wang Z, Song J, Milne TA, Wang GG, Li H, et al. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell. 2010; 141: 1183-1194. PubMed: https://pubmed.ncbi.nlm.nih.gov/20541251/
- Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci U S A., 2003; 100: 8342-8347. PubMed: https://pubmed.ncbi.nlm.nih.gov/12829790/