
More Information
Submitted: September 28, 2023 | Approved: October 13, 2023 | Published: October 14, 2023
How to cite this article: Tichon A, Eitan E, Tsory S, Beit-Yanai E, Priel E. The Neuroprotective Role of TERT Influences the Expression of SOD1 in Motor Neurons and Mouse Brain: Implications for fALS. J Neurosci Neurol Disord. 2023; 7: 113-125.
DOI: 10.29328/journal.jnnd.1001085
Copyright License: © 2023 Tichon A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Telomerase, H2O2; Mitochondria; Catalase; SOD1; TERT; TERT KO mice

The Neuroprotective Role of TERT Influences the Expression of SOD1 in Motor Neurons and Mouse Brain: Implications for fALS
Ailone Tichon1, Erez Eitan1, Sylvia Tsory1, Elie Beit-Yanai2 and Esther Priel1,3*
1The Shraga Segal Department of Immunology and Microbiology, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel
2The Department of Clinical Biochemistry and Pharmacology, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel
3Neuromagen Pharma Ltd. High Tech Park, Beer-Sheva, Israel
*Address for Correspondence: Esther Priel, The Shraga Segal Department of Immunology and Microbiology, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel, Neuromagen Pharma Ltd. High Tech Park, Beer-Sheva, Israel, Email: [email protected]
Amyotrophic lateral sclerosis (ALS) disease is characterized by degeneration of motor neurons and elevation of brain oxidative stress. Previous studies demonstrated the neuroprotective effects of Telomerase reverse transcriptase (TERT) from oxidative stress. We showed that increasing TERT expression in the brain of the Tg hSOD1G93A mouse ALS model attenuated the disease pathology and increased the survival of motor neurons exposed to oxidative stress. How TERT increased the survival of motor neurons exposed to oxidative stress is not yet clear. Here we investigated the consequence of TERT depletion in motor neuron cells under normal and oxidative stress conditions and in mouse brains of TERT knockout mice, on the expression and activity of SOD1 and catalase enzymes. Depletion of mouse TERT caused mitochondrial dysfunction and impaired catalase and SOD1 activity. Compensation with hTERT restored the activity of SOD1. SOD1 expression increased in the brain of TERT KO and in ALS mice and decreased in the brain of WT mice treated with telomerase-increasing compounds. We suggest that the ability of TERT to protect neurons from oxidative stress affects the expression and activity of SOD1, in a TERT-dependent manner, and supports the notion of TERT as a therapeutic target for neurodegenerative diseases like ALS.
Amyotrophic Lateral Sclerosis (ALS) known also as Lou Gehrig`s disease is a paralytic disorder that is characterized by degeneration of the upper motor neurons in the motor cortex and lower motor neurons in the brainstem and spinal cord that causes progressive denervation of voluntary muscles. This is a progressive and lethal disease with a low life expectancy of two to five years. The cause of sporadic ALS is yet unknown. The mechanisms involved in motor neuron degeneration are multifactorial and complex. The heterogeneity of ALS suggests that different mechanisms may play prominent roles in individual patients. ALS is a complex disease in 10% - 21% of patients it demonstrates a monogenic while in the majority of affected individuals, an interaction of multiple genetic and environmental risk factors was detected [1]. Accumulating evidence suggests oxidative stress as potential pathogenesis of the disease: 1. Increased oxidative damage is found in vast regions in the brains of ALS patients and in mouse disease models [1-3], including neurons and glia cells [4]. 2. Reactive Oxygen Species (ROS) was demonstrated to mediate the damage of glutamate excitotoxicity, mitochondrial dysfunction, mutant SOD1 protein aggregation, and abnormal accumulation of neurofilaments [5-8]; all of them were suggested to be ALS pathogenesis factors. In addition, human Telomerase Reverse Transcriptase (hTERT) expression level was significantly lower in ALS patients and was correlated either to p53 mRNA expression or p21 expression, suggesting that a decrease in telomerase could be a pathogenic contributor to neurodegeneration in ALS [9].
Telomerase is a ribonucleoprotein complex, which functions in telomeres maintenance. The active enzyme consists of Telomerase Reverse Transcriptase (TERT), an RNA subunit (TERC), and several additional proteins that are important for telomerase assembly, stability, and trafficking into the telomere [10-12]. TERT is mainly active in highly proliferated cells (e.g., antigen-stimulated lymphocytes, germ cells, embryonic cells, and most cancers). In somatic cells TERT expression is tightly regulated therefore, somatic cells usually do not demonstrate telomerase activity (mechanisms of TERT regulation are summarized in [13]. Emerging evidence indicates that low levels of TERT expression are observed in somatic cells such as Purkinje cells in the brain [14], bone marrow mesenchymal stromal cells [15], breast tissues [16], epithelial cells [17] and fibroblasts [18]. In the last decade, we and others demonstrated the neuroprotective effects of TERT in mouse brain [19-23]. In addition, accumulating evidence indicates that TERT possesses additional non-telomeric activities. Under oxidative stress, TERT migrates from the nucleus into the mitochondria. This migration is followed by an improved coupling of the Mitochondrial Membrane Potential (MMP) with ATP production and by inhibition of the mitochondrial apoptotic pathway [24,25]. The mechanism for these activities of TERT is not completely understood. It has been shown that TERT lacking reverse transcriptase activity can still protect mouse and human cells, thus the protective effect of TERT is not necessarily related to its ability to maintain telomeres [26]. In addition, TERT did not improve base excision repair in the mitochondria under oxidative stress but increased the level of MnSOD protein [27]. It was previously shown in our lab that overexpression of human TERT (hTERT) reduced the sensitivity of NSC-34 cells, which are a mouse motor neuron-like cell line, to oxidative damage [14], indicating hTERT ability to function in mouse cells. We also showed that increasing telomerase activity and expression in the brain by specific compounds (AGS) designed by our laboratory resulted in attenuation of the ALS disease pathology in the Tg SOD1G93A mouse model [21]. We also showed that increasing telomerase expression and activity by AGS compounds protect hippocampal neurons from amyloid beta toxicity by enhancing the expression of neurotrophins and plasticity-related genes [19].
One may speculate that the therapeutic benefits that telomerase confers for ALS disease are mediated by its ability to protect motor neurons from oxidative stress. Here we examined this hypothesis and the way in which TERT mediates this effect in motor neurons like cells (NSC-34) that were depleted from the mouse TERT and in some experiments were compensated with hTERT following the depletion of mouse TERT. In addition, we also examined the effect of TERT on the expression of oxidative stress enzymes (SOD1 and catalase) in vivo in the brains of WT and TERT KO mice. The expression of these enzymes was also examined in the brain of hSOD1 Tg mice the model for ALS.
These results indicate that the ability of mouse TERT to protect mouse motor neuron cells from oxidative stress is in part mediated by its effect on the expression and activity of antioxidant enzymes such as SOD1 and catalase and that human TERT can partially compensate for mouse TERT depletion.
Cell cultures
Mouse NSC-34 cell line: Mouse NSC-34 cells, a motor neuron-like cell line [28], was kindly received from Dr. Daniel Offen, Tel Aviv University, Israel. The cells were grown in medium DMEM (4.5 gm/l D-Glucose) supplemented with 1% L-Glutamine, 1% penicillin-streptomycin (Biological Industries, Beit Haemek, Israel), and 10% FCS (Gibco, Carlsbad, CA, USA) at 37 ºC and 5% CO2.
Plasmids
For TERT manipulation in NSC-34: TRCN0000070968-72 Vectors: pLKO.1-puro containing mTERT shRNA (Sigma, Rehovot, Israel) was screened for the efficacy of mTERT depletion. Then, a combination of both TRCN0000070971 and TRCN0000070969 was used for the depletion of mTERT in the NSC-34 cells. For hTERT over-expression we used a pWZL-Blasticidin vector containing flag-HA- hTERT and Scrambled shRNA (Addgene, Cambridge, MA, USA)
Lentivirus production
Packaging of the mTERT shRNAs TRCN0000070971 and TRCN0000070969 vectors was performed in GP2-293 packaging cells (Clontech, Mountain View, CA, USA) as previously described [29] using pCMVVSV-G, pMDL.gp.RRE and pRSV-Rev plasmids kindly provided by Dr. A. Chen (Weizmann Institute of Science, Rehovot, Israel).
NSC-34 gene manipulation
For mTERT down-regulation in the NSC-34 cell line, cells (106) were grown in a T-25 flask and were incubated with lentivirus containing both mTERT shRNAs TRCN0000070971 and TRCN0000070969 for 48h, following growth in medium containing 10 µg/ml puromycin (Sigma, Saint Louis, MO, USA) for additional 3 - 5 days. For hTERT overexpression, the NSC-34mTERT-shRNA cells, which were confirmed for mTERT down-regulation, were transfected with pWZL-Blasticidin containing flag- HA-hTERT using the jetPrime Polyplus Transfection reagent (POLYPLUS- TRANSFECTION SA, Cedex, France) according to manufacturer’s protocol for 24h.
Cells were then grown in a medium containing 1 – 5 µg/ml Blasticidin (Sigma, Saint Louis, MO, USA) for an additional\3 - 5 day.
Preparation of protein extract
Cells were washed with PBS to remove the medium. Then, the cells were incubated with CHAPS buffer lysis, containing 10mM TRIS HCl pH-7, 1 mM MgCl2, 1mM EDTA, 0.1 mM PMSF (phenyl-methylsulfonyl), 0.5% CHAPS (3[(3 Cholamidopropyl) dimethylammonio]-propanesulfonic acid) and 10% Glycerol for 30 minutes in 4 ºC. Then, the cells were centrifuged for 30 minutes at 13000 RPM at 4 ºC. The supernatant was collected, and the total protein concentration was determined by BIO- -Rad protein assay kit (Bio-Rad Laboratories).
Telomerase activity
TRAP assay: was performed as previously described [30]. In brief, protein extract (at the indicated amount) was incubated with TS primer (5`AATCCGTCGAGCAGAGTT-3’) for 45 min at 30 oC followed by PCR assay with [α-P32] dCTP using ACX primer: (5’- GCG CGG CTT ACC CTT ACC CTT ACC CTA ACC-3’). Internal standard primers used as a control: IS primer (5’AAT CCG TCG AGC AGA GTT AAA AGG CCG AGA AGC GAT-3’) and ISR primer (5’-ATC GCT TCT CGG CCT TTT-3’). For the detection of telomerase products of hMSC 32 PCR cycles were used, and the internal standard primers were diluted to a concentration of 5 x 10-15M. The PCR products were labeled by [α-P32] dCTP and separated on 12.5% polyacrylamide gel. The radioactive labeled products were detected by a Phosphoimager (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot analysis
Antibodies: SOD1 was measured using anti-SOD1 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). β-actin was measured using an anti-β-actin antibody (ICN, Irvine, CA, USA). Protein extract was prepared and analyzed by Western blot assay as previously described [31,32] using the various antibodies. The immune complexes were detected by enhanced chemiluminescence (ECL) (Biological Industries., BeitHaemek, Israel). Densitometric analysis was performed using the EZ-Quant software (EZ-Quant, Rehovot, Israel).
Cell survival under oxidative stress
Cells were plated as triplicate in 96-well plates at a density of 20000 cells/well and were exposed to various concentrations of H2O2 (10 - 100 nM) for 1h, in serum-free medium. The medium was replaced with fresh serum-containing medium for 24h. Cell cytotoxicity was measured by the Neutral Red assay.
Detection of antioxidant properties and cellular ROS accumulation
Intracellular ROS formation and stabilization due to exposure of cells to H2O2 was detected using the reagent 2’-7’-Dichlorodihydrofluorescein diacetate (Sigma, Saint Louis, MO, USA), as previously described [33,34]. Briefly, Cells were cultured overnight in triplicate in a 96-well plate at a density of 20000 cells/well). The following day, the cells were incubated with 0, 20, and 40 µM of H2O2 for 30 minutes at 37 ºC and 5% CO2. The medium was removed, cells were washed with PBS and a normal growth medium containing DCFH-DA was added. Using TECAN Infinite® 200 PRO microplate readers the cells were measured every minute for 45 minuts long at excitation 485 nM, emission 520 nM.
Measurement of mitochondrial membrane potential (MMP)
The different NSC-34 cells were cultured as triplicates in 96-well plates at a density of 20000 cells/well overnight. Day after, cells were incubated with Tetra-Methyl- Rhodamine-Methyl-Ester (TMRM) (Sigma, Saint Louis, MO, USA) at 50 µM for 45 min in medium DMEM without phenol red (Biological Industries, Beit Haemek, Israel). Then, cells were exposed to 0 and 20 µM H2O2 for 30 minutes in a serum-free medium. TMRM emission at 573 nm was measured using Eliza (Bio-Rad Laboratories, Hercules, CA, USA).
Measuring the proton motive force
Measurement of proton motive force was performed as previously described [35]. Briefly, the different NSC-34 cells were cultured (3 x 105 cells) in fluoro-dish (NBT, Jerusalem, Israel) overnight. Day after, the cells were incubated with TMRM [50 µM] supplemented to medium DMEM without phenol red for 45 min (Biological Industries, Beit Haemek, Israel). At the indicated time, 1 µM of Carbonylcyanide-4- (trifluoromethoxy)-phenylhydrazone (FCCP) was added to the cells and the rate of decay in TMRM (fluorescent intensity reduction/min, illumination at 514 nm and detection at 570 nm) was visualized and analyzed by the confocal microscopy system FV1000 (Olympus, Tokyo, Japan).
Measurement of ATP concentration
ATP concentration in the different NSC-34 cells was measured with the “ATP Bioluminescence Assay Kit HSII” (Roche, Basel, Switzerland), according to the manufacturer’s recommended protocol, using the Glomax 20/20 luminometer (Promega, Fitchburg, Wisconsin, USA).
Quantification of gene expression qRT-PCR
Total RNA from the different NSC-34 cells was prepared as previously described [36] and then was transcribed to cDNA with the “Revert Aid First Strand Synthesis Kit” (Thermo Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s instructions. Then, quantitative Real-Time PCR was performed using “Fast Start Universal SYBR Green Master” (Roche, Basel, Switzerland) with the 7500 Real-time PCR System (Applied BioSystems, Carlsbad, California, USA) for the following primers:
| β-actin | Fw: 5’-GCA GCT CAG TAA CAG TCC GCC T-3’ Rv: 5’-TGG CCT CAC TGT CCA CCT TCC A-3’ |
| SOD1 | Fw: 5’-TCC GTC GGC TTC TCG TCT TG-3’ Rv: 5’-TCA CCG CTT GCC TTC TGC TC-3’ |
| Catalase | Fw: 5’-CTT CAG GGC CGC CTT TTT GC-3’ Rv: 5’-ATA GTT GGG GGC ACC CT-3’ |
| mTERT | Fw: 5’-GAA AGT AGA GGA TTG CCA CTG GC-3’ Rv: 5’-CGT ATG TGT CCA TCA GCC AGA AC-3’ |
| hTERT | Fw: 5’-GCC GAT TGT GAA CAT GGA CTA CG-3’ Rv: 5’-GCT CGT AGT TGA GCA CGC TGA A-3’ |
Antioxidants enzymatic activity
Superoxide dismutase1 (SOD1): Enzymatic activity was evaluated indirectly using the nitro blue tetrazolium assay as previously described [37]. Briefly, the reaction medium and the required amount of protein extract (10 µg – 15 µg) were illuminated for 10 minutes at 25 °C, and absorbance at 560 nm was subsequently measured by microplate reader 680 (Bio-Rad Laboratories, Hercules, CA, USA).
Catalase: Reaction was initiated by the addition of 60 mM H2O2 to protein extract (10 µg - 15µg) in phosphate buffer. At fixed time intervals, aliquots were removed and quenched by addition into a mixture of 0.6 N H2SO4: 10mM FeSO (4:1, v/v). After all samples had been collected, color was developed by the addition of 2.5 M KSCN. Color intensity was measured at 490 nm by microplate reader model 680 (Bio-Rad, Hercules, CA, USA).
In vivo studies
Mice: All animal experiments were approved by the animal ethics committee at Ben-Gurion University (IL-07-06-14 and IL-11-09-2018C.) Tert heterozygous (B6.129S-Terttm1Yjc/J) and their WT counterparts, male and female mice were purchased from Jackson Laboratory. TERT KO mice were established and identified by genotyping and the lack of TERT expression and activity in the brain and other organs. WT, TERT KO mice (generation 2 and generation 3) and ALS mice (B6SJL- Tg (SOD1*G93A)1Gur/J) at the age of 3 - 4 months were sacrificed and the brain was removed as previously described by us [14,19,21].
AGS treatment: WT Mice (C57BL/6) (3 months old) were subcutaneously injected with AGS-534 (6 mg/kg) or vehicle (DMSO) as we previously described [13,18,19]. Twelve hours post treatment mice were sacrificed and the brain was removed and subjected to RNA extraction and gene expression procedures using the appropriate DNA primers as we previously described [14,19,21].
Statistical analysis
quantitative analysis results are the mean of at least 3 independent experiments ± standard deviation. The significance of the results was performed using the student’s T-test or One-Way ANOVA by the GraphPad InStat software (GraphPad Software, La Jolla, CA, USA).
NSC-34 cells were depleted of mTERT and compensated with hTERT
To deplete mTERT in the NSC-34 wild-type cells (NSC34WT) the effect of a series of putative mTERT shRNA vectors TRCN68 - 72 on mTERT expression and telomerase activity was examined. TRCN69, 70 and 71 vectors efficiently reduced mTERT expression and telomerase activity (Supplemental 1).
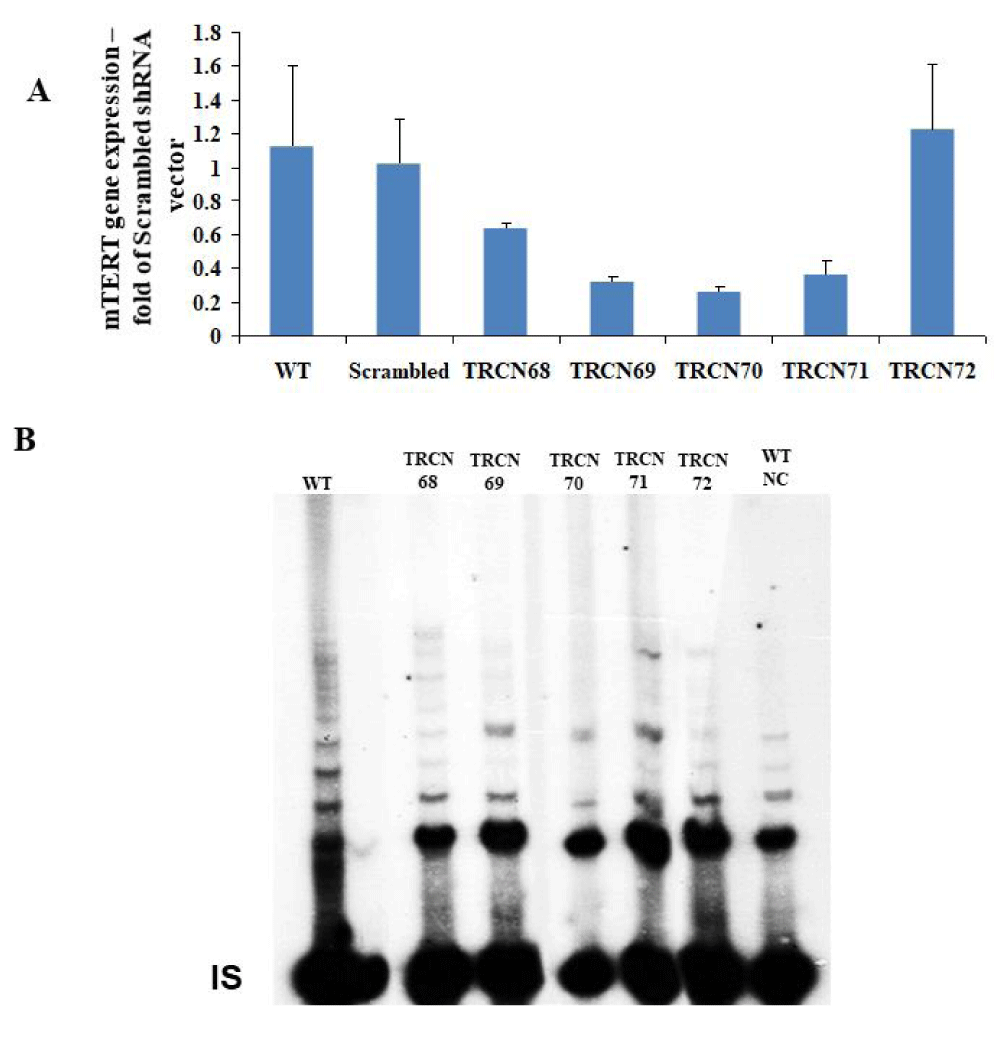
Suplemental Figure 1: Screening of mTERT shRNA vectors for the depletion of TERT expression in NSC-34 cells. A. The effect of mTERT shRNA TRCN vectors (purchase from Sigma) on mTERT gene expression relative to β-actin in NSC-34 cells. Gene expression was measured using qRT-PCR and is represented in the graph as the mean of 3 different measurements ± SD. The results are fold of Scrambled shRNA vector. B. The effect of transfected mTERT shRNA TRCN vectors to WT NSC-34 cells on telomerase activity using the radioactive TRAP assay. Symbols: WT – Wild Type, NC – Negative Control by boiling the WT protein extract, IS- internal standard
Then, a combination of TRCN 69 and TRCN71 vectors was employed to generate mTERT knocked-down cells (NSC34mTERTshRNA) and down regulation of mTERT was observed by detecting telomerase activity (Fig. 1A, compare lane 2 to lane 3) and by measuring mTERT gene expression (Figure 1B). Next, hTERT was introduced to the NSC34mTERT-shRNA (NSC34mTERT-shRNA hTERT), which result in a restoration of telomerase activity, although in a different pattern (Figure 1A, compare lane 5 to lane 2 and 3), and was confirmed by real time PCR with human TERT specific primers (Figure 1C). Impaired growth and proliferation capacity were observed in the NSC34mTERT-shRNA compared to the NSC34WT cells (Figure 1D). Interestingly, no improvement in proliferation and growth capacity was observed in the NSC34mTERT-shRNA hTERT cells compared to the NSC34mTERT-shRNA cells (Figure 1D), suggesting that hTERT cannot replace mTERT for this purpose.
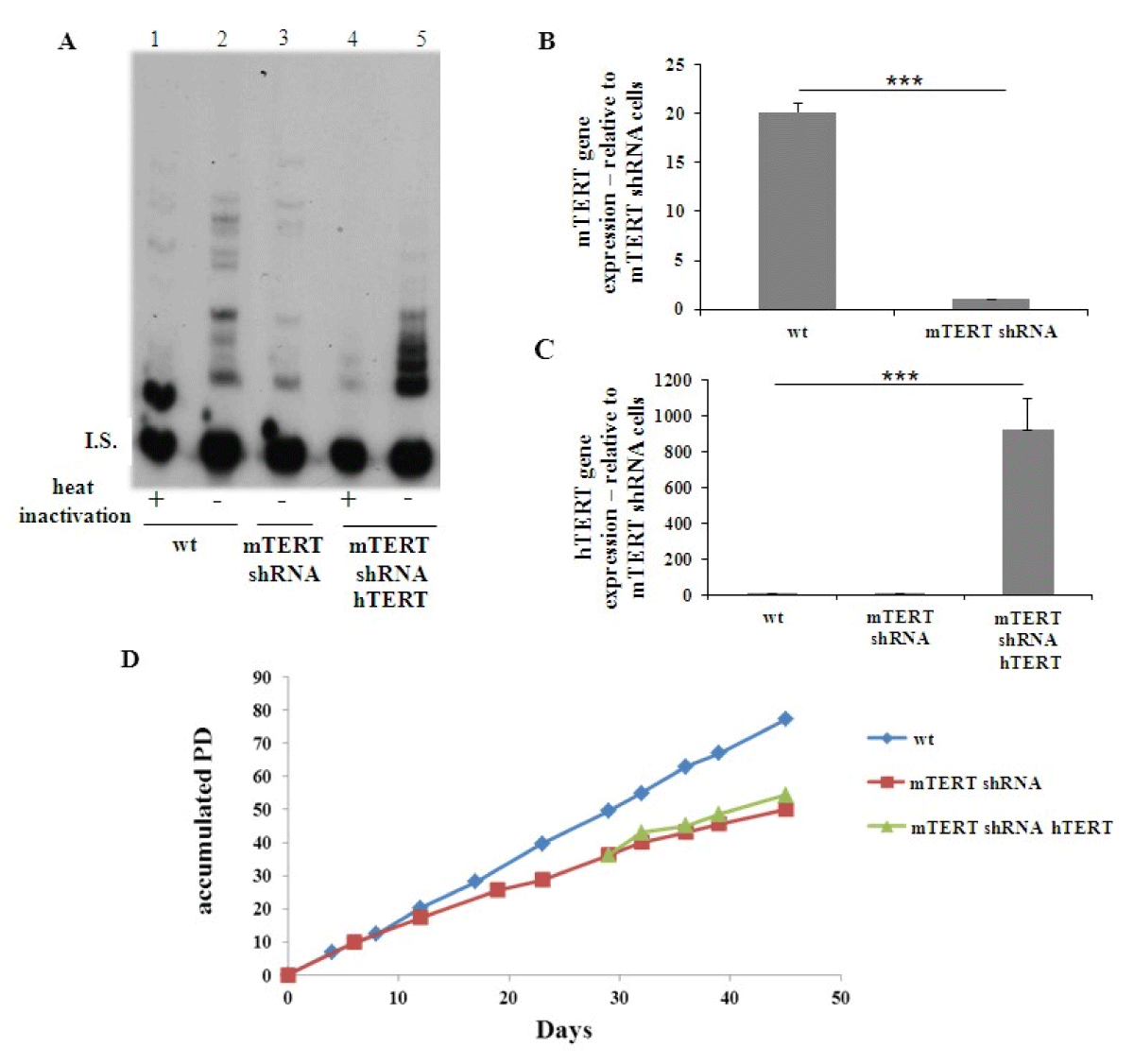
Figure 1: Depletion of mTERT expression and activity in NSC-34 cells and restoration of the activity and expression by hTERT. NSC-34 cells (NSC-34WT) were depleted of mTERT (NSC-34mTERT shRNA), and samples were transfected with hTERT expressing vector (NSC-34mTERT shRNA hTERT). (A). The activity of telomerase detected by TRAP assay in the NSC-34WT cells (lanes 1 and 2), NSC-34mTERT shRNA (lane 3), and NSC-34mTERT shRNA hTERT (lanes 4 and 5). I.S. – Internal standard Negative control – heat inactivation of the protein extract. B and C. Quantitative gene expression analysis by TR-PCR of mTERT (B) in the NSC-34WT and NSC-34mTERT shRNA cells (n = 3 independent experiments); and of hTERT (C) in NSC-34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT (n = 3 independent experiments). The results are normalized to β-actin gene expression. Significance – Student’s t-test. ***p < 0.001. D. A graph representing the accumulated PD the cells undergo with time (n = 2 independent experiments).
TERT protects NSC-34 from the insults of H2O2 oxidative stress
To examine the possible TERT protective effect from oxidative stress in NSC-34 cells, the various NSC-34 transfected cells (NSC-34WT, NSC-34mTERT shRNA, and NSC- 34mTERT shRNA hTERT) were treated with increasing doses of H2O2 (0 nM – 100 nM) and the CC50 (the dose that kills 50% of the cells) was determined (Figure 2A). The H2O2 CC50 in NSC-34WT, NSC-34mTERT-shRNA hTERT, and NSC-34mTERT-shRNA cells was 34 µM, 25 µM, and 18 µM, respectively (Figure 2A), indicating that TERT depletion increased the sensitivity of NSC-34 cells to H2O2, and restoration of telomerase expression by hTERT partially protected the cells from oxidative stress.
The accumulation of intracellular ROS was measured using the DCFH-DA reagent. In the absence of H2O2, no differences in the levels of intracellular ROS were observed between the cells (Figure 2B). However, in the presence of 20 µM or 40 µM H2O2 the levels of intracellular ROS were higher in the NSC-34mTERT-shRNA cells, while in the NSC34mTERT-shRNA hTERT cells ROS levels decreased to the levels observed in the NSC-34WT cells (Figure 2C-D).
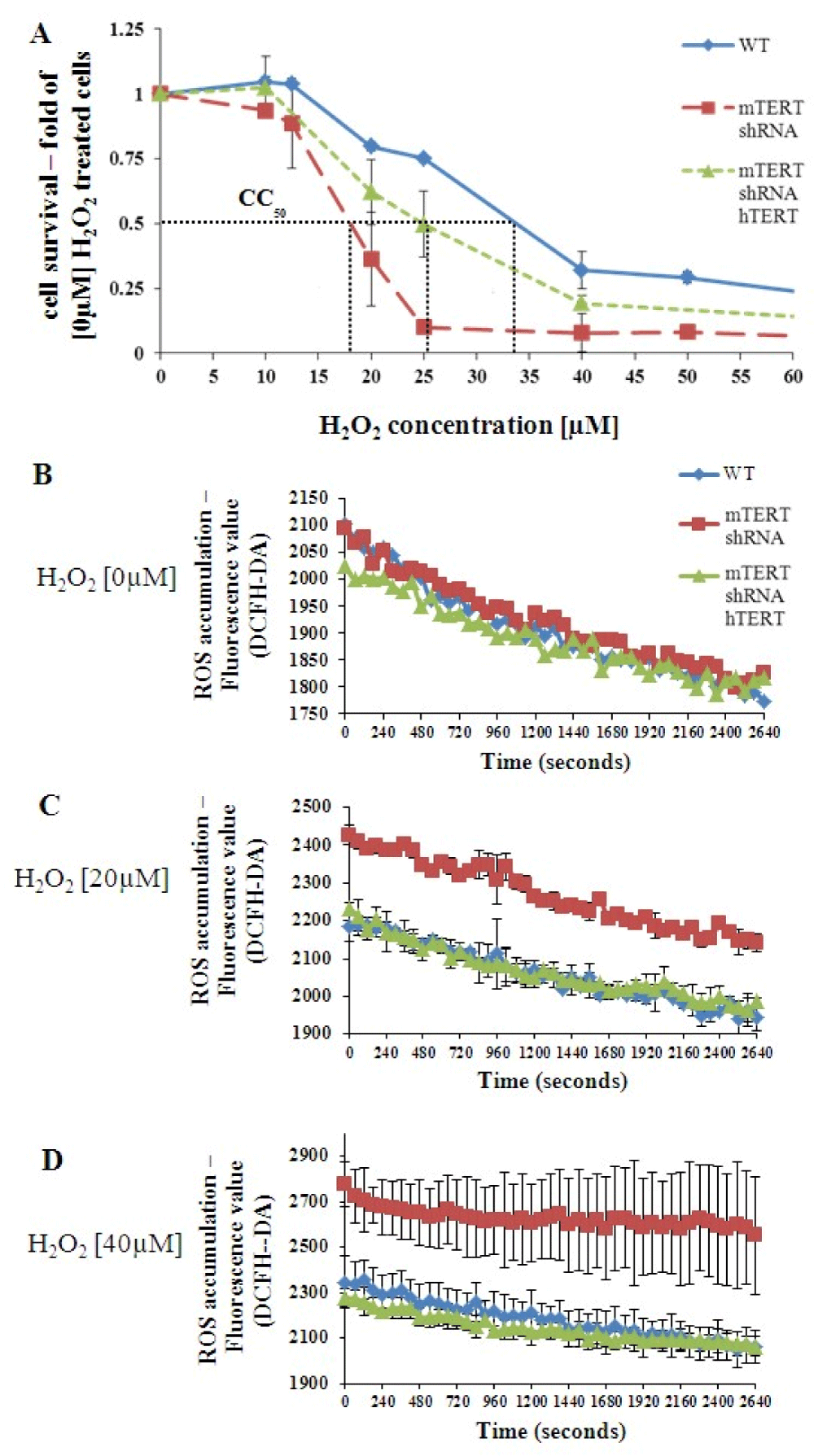
Figure 2: TERT protects NSC-34 from the H2O2–induced oxidative stress. (A). A graph representing the cytotoxic effect of gradually increasing doses of H2O2 on survival of NSC-34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT cells. CC50 = H2O2 concentration is required for killing 50% of cells. B-D. Measurement of intracellular ROS accumulated in the NSC-34WT, NSC-34mTERT shRNA, and NSC- 34mTERT shRNA hTERT cells exposed to H2O2 0 µM (B), 20 µM (C), and 40 µM (D). The results are means of at least 3 different experiments.
Depletion of mTERT affected Mitochondrial Membrane Potential (MMP) and intracellular ATP levels
The by-product of the ATP generation process in the mitochondria is the formation of ROS. Therefore, we investigated the effect of TERT on the mitochondrion’s functionality under H2O2-induced oxidative stress.
The MMP is decreased following exposure to H2O2 [38]. Using TMRM reagent, the effect of TERT on intracellular MMP was determined. In the absence of H2O2, the NSC-34mTERT-shRNA, and NSC-34mTERT-shRNA hTERT cells demonstrated a higher MMP compared to NSC-34WT cells (1.61 ± 0.008 fold of WT, p < 0.001 and 1.68 ± 0.11 fold of WT, p < 0.001, respectively; Figure 3A). In the presence of 20 µM H2O2, the MMP in the NSC-34WT cells increased by 1.31 ± 0.06 fold, while in the NSC-34mTERT-shRNA and NSC-34mTERT-shRNA hTERT cells, it decreased to the level observed in the NSC-34WT cells (1.23 ± 0.14 fold, a reduction of 23%, p < 0.05, and 1.17 ± 0.12 fold, a reduction of 30%, p < 0.05, respectively). These results are not compatible with a previous report showing that in human fibroblasts, in non-oxidative stress conditions, the over-expression of hTERT resulted in an increased MMP compared to their control cells [24]. Therefore, the Proton Motive Force (PMF) was evaluated by measuring the rate of MMP depolarization using the protonophore Carbonyl-cyanide-4- (trifluoromethoxy)-phenylhydrazone (FCCP). In the NSC-34mTERT-shRNA and NSC-34mTERT-shRNA hTERT, the rate of MMP depolarization was significantly faster than in the NSC-34WT cells (1.31 ± 0.06-fold, p < 0.05, and 1.51 ± 0.1, p < 0.01, respectively; Figure 3B, indicating higher PMF in the mTERT-depleted cells compared to the WT cells. PMF is used for the generation of ATP thus we examined the ATP levels. NSC- 34mTERT shRNA and NSC-34mTERT shRNA hTERT have decreased ATP levels compared to NSC-34WT cells (0.58 ± 0.25-fold, p < 0.05, and 0.32 ± 0.24-fold, p < 0.01 respectively) (Figure 3C). These results demonstrate significant changes in mitochondria functionality and cell energy levels due to depletion of mTERT. However, the introduction of hTERT did not compensate for the depletion of mTERT, suggesting the species-dependent function of TERT in the mitochondria.
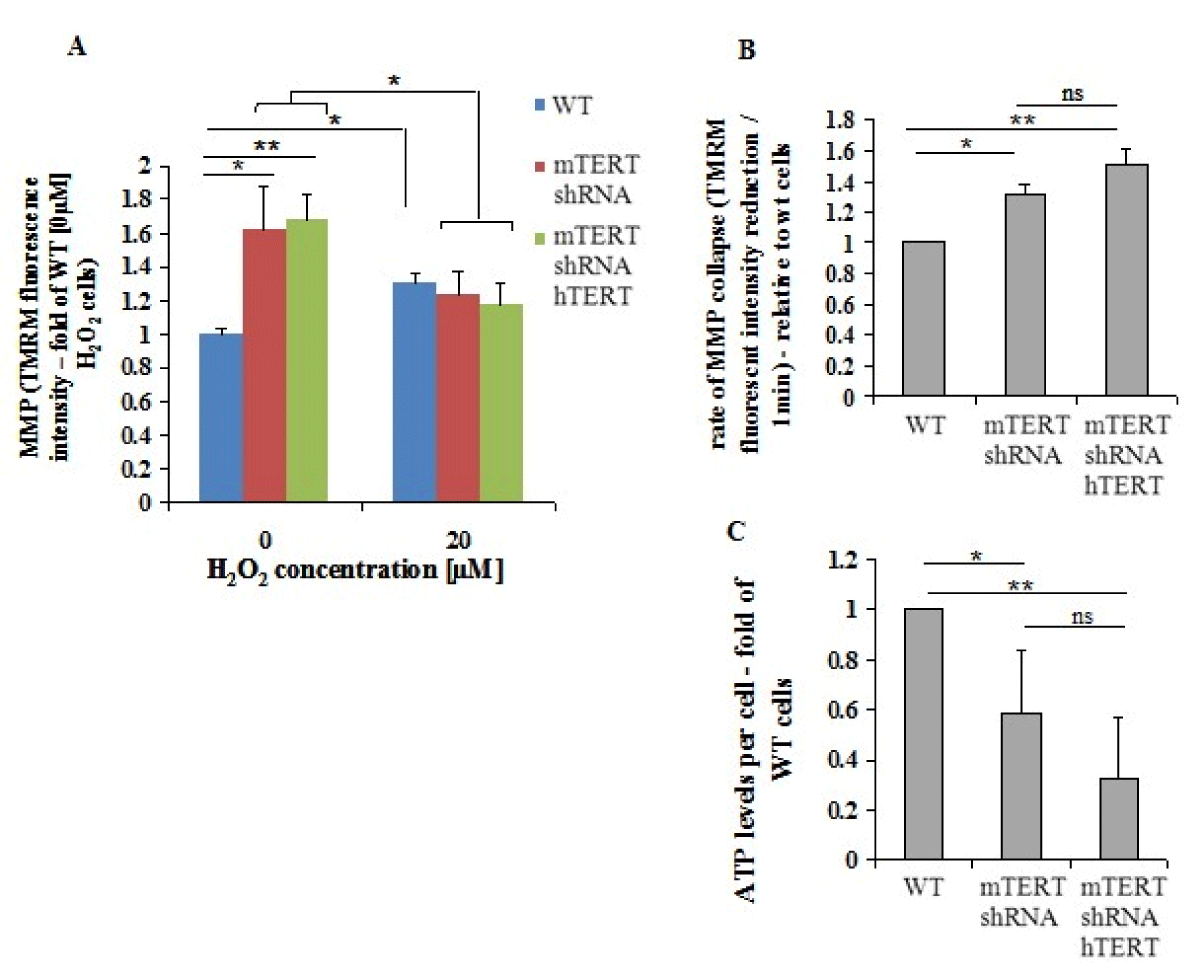
Figure 3: TERT affects MMP and ATP levels. A. The MMP was measured in the presence of 0µM and 20µM H2O2 in the NSC- 34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT using TMRM reagent. B. The PMF in the NSC-34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT cells was demonstrated by measuring the MMP depolarization rate, by quantifying the reduction rate of TMRM fluorescent intensity due to the addition of the protonophore FCCP. C. ATP levels in the NSC-34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT quantified using the ATP Bioluminescence Assay Kit HSII (Roche, Basel, Switzerland). The results were normalized to a number of cells. All the results are represented as folds of the NSC-34WT cells. The results are means of n = 3 different experiments, significance by one-way ANOVA and Tukey’s post hoc analysis. *p < 0.05, **p < 0.01.
TERT affects the expression and activity of Catalase and Superoxide dismutase 1 under normal or oxidative stress conditions
To further investigate the way TERT expression protects motor neuron cells from oxidative stress we investigated the fate of antioxidant enzymes such as Superoxide Dismutase1 (SOD1) and Catalase, in TERT depleted and TERT knock-in cells.
In the absence of H2O2, the NSC-34mTERT shRNA exhibited an increased expression of SOD1 (1.36 ± 0.13-fold, p < 0.01) and Catalase (1.94 ± 0.37-fold, p < 0.05) genes relative to NSC-34WT cells (Figure 4A). However, in the NSC-34mTERT shRNA hTERT cells, the expression of Catalase was restored to the levels observed in the NSC-34WT cells (Figure 4A), while the expression of the SOD1 gene was even lower than the levels of the NSC34WT cells (0.43 ± 0.24 fold of WT, p < 0.001; Figure 4A). This effect was confirmed by the examination of SOD1 protein levels in the different cells, revealing a reduction in SOD1 levels in the NSC-34mTERT shRNA hTERT cells relative to the NSC- 34WT cells. Nevertheless, although an increase in SOD1 mRNA was detected in the NSC-34mTERTshRNA compared to the NSC-34WT cells, no effect on the levels of SOD1 protein was observed (not shown). Exposures of cells to H2O2 [20 µM] resulted in an elevated expression of SOD1 and Catalase genes in the NSC-34WT cells (1.3 ± 0.05- fold, p < 0.01; and 1.5 ± 0.17-fold, p < 0.01, respectively) and in the NSC-34mTERTshRNA cells (2.21 ± 0.15-fold, p < 0.001; and 3.41 ± 0.3-fold, p < 0.001, respectively) relative to the NSC-34WT cells untreated with H2O2 (Figure 4B).
To examine whether the differences in the gene expression are compatible with the activity, whole-cell proteins were extracted and the activities of catalase and SOD1 in the presence and absence of H2O2 were measured. Without H2O2 exposure, the Catalase activity in the NSC-34mTERT shRNA and in the NSC-34mTERT shRNA hTERT cells was higher than in the NSC-34WT cells (3.7 ± 1.1-fold, p < 0.001; and 1.5 ± 0.14-fold, p < 0.05, respectively; Figure 4C). Under H2O2 [20 µM] exposure, the catalase activity in the NSC-34mTERT shRNA and in the NSC-34mTERT shRNA hTERT was significantly reduced compared to that observed in the NSC-34WT cells (0.43 ± 0.2-fold, p < 0.01; and 0.56 ± 0.07-fold, p < 0.05, respectively; Figure 4C). It was reported that sub-lethal doses of ROS increased cellular catalase activity as an intrinsic protective mechanism. Indeed, exposure of the cells to oxidative stress increased the Catalase activity in the NSC-34WT cells (by 5.5 ± 2.4-fold, p < 0.001), while no significant induction was observed in the NSC34mTERT shRNA cells (Figure 4C). However, a partial compensation was observed in the NSC-34mTERT shRNA hTERT cells, as H2O2 exposure resulted in increased catalase activity by 2.1 ± 0.3-fold, p < 0.05, relative to cells untreated with H2O2 (Figure 4C).
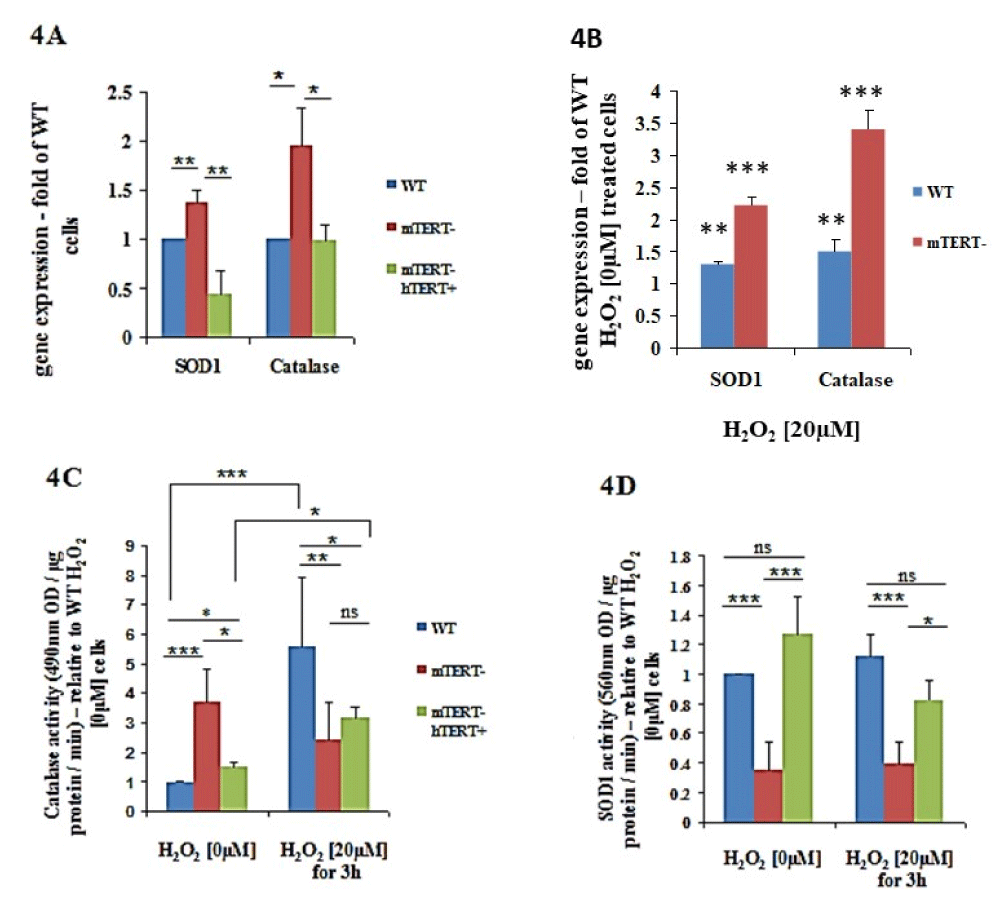
Figure 4: The effects of TERT on expression and activity of antioxidant genes SOD1 and Catalase in normal and oxidative stress conditions. A. Quantification of the expression of SOD1 and Catalase genes normalized to β-actin in the NSC-34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT performed by qRTPCR. The results are represented as fold of the NSC-34WT cells (without H2O2) and are means of n = 5 different experiments. Significance by one-way ANOVA and Tukey’s post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001. B. Quantification of the expression of SOD1 and Catalase genes normalized to β-actin in the NSC-34WT, NSC34mTERT shRNA cells exposed to 20 µM H2O2. The results are represented as a fold of the NSC- 34WT cells (without H2O2) are means of n=5 different experiments. C and D. Quantification of the antioxidant activity of Catalase (C) and SOD1 (D) in the NSC- 34WT, NSC-34mTERT shRNA, and NSC-34mTERT shRNA hTERT cells treated with or without 20 µM H2O2. All the results are represented as folds of the NSC-34WT cells. The results are means of 6 different experiments. Significance by one-way ANOVA and Tukey’s post hoc analysis. *p < 0.05, **p < 0.01, ***p < 0.001
The effect of TERT on the activity of SOD1 was examined, and under non-oxidative stress conditions, a significant reduction in SOD1 activity (0.34 ± 0.19 fold of WT, p < 0.001; Figure 4E) was observed in the NSC-34mTERT shRNA cells. In contrast, the SOD1 activity in NSC-34mTERT- hTERT+ significantly increased compared to the NSC-34mTERT shRNA cells (3.7 ± 0.2 fold higher, p < 0.001; Figure 4D). Under oxidative stress conditions, a similar pattern of activity was detected: SOD1 activity in the NSC-34mTERT- cells was decreased by 0.34 ± 0.15 fold of the NSC-34WT, p < 0.001, and the activity in the NSC34mTERT shRNA hTERT was increased by 2.12 ± 0.14 fold of NSC-34mTERT shRNA cells, p < 0.05 (Figure 4D).
SOD1 and catalase gene expressions in mouse brains decreased following an increase in TERT expression by AGS compound and increase in TERT knockout mouse brain The endogenous SOD1 expression increased in the brain of ALS mice and TERT KO mice and decreased in AGS-treated WT mice.
TERT influences the expression of catalase and SOD1 in mouse brain
To examine the effect of increased TERT expression in vivo in mouse brain we treated the WT mice with a single injection of AGS -534, a TERT increasing compound [15,18,19]. RNA brain extracts were prepared 12 hr. after AGS treatment and SOD1 and Catalase gene expressions were detected by Real-Time PCR using the appropriate primers. As seen in Figure 5A Increasing TERT expression in the brain by pharmaceutical compounds significantly decreased the expression of SOD1 to 78 ± 7.09%, p = 0.001, and catalase to 67 ± 8.3% p = 0.004. To determine the effect of TERT knockout on the expression of SOD1 in the mouse brain we prepared RNA extracts from the brain derived from TERT KO generation 2 and generation 3 mice and from their WT counterpart and examined the expression of SOD1 gene. As can be seen in Figure 5B the expression of SOD1 significantly increased in the brain of TERT KO mice to 121.1% ± 0.01%, p = 0.0012 for G2, and to 122 ± 0.02%, p = 0.00192 for G3 compared to that found in the WT counterpart. The results obtained in the mouse brain are compatible with the motor neuron cell culture data showing the impact of TERT on SOD1 and catalase gene expressions. Finally, since elevation of oxidative stress in the brain and spinal cord, was suggested as one of the possible factors for motor neuron degeneration in ALS disease, we examined the expression of SOD1 and catalase in the brain of ALS mouse model (hSOD1G93A Tg mice) at the symptomatic stage. The results revealed a significant increase in SOD1 by 2.4-fold (Figure 5C) and catalase by 3-fold (Figure 5D) compared to the WT counterpart suggesting a high oxidative stress situation in the brain of ALS mice.
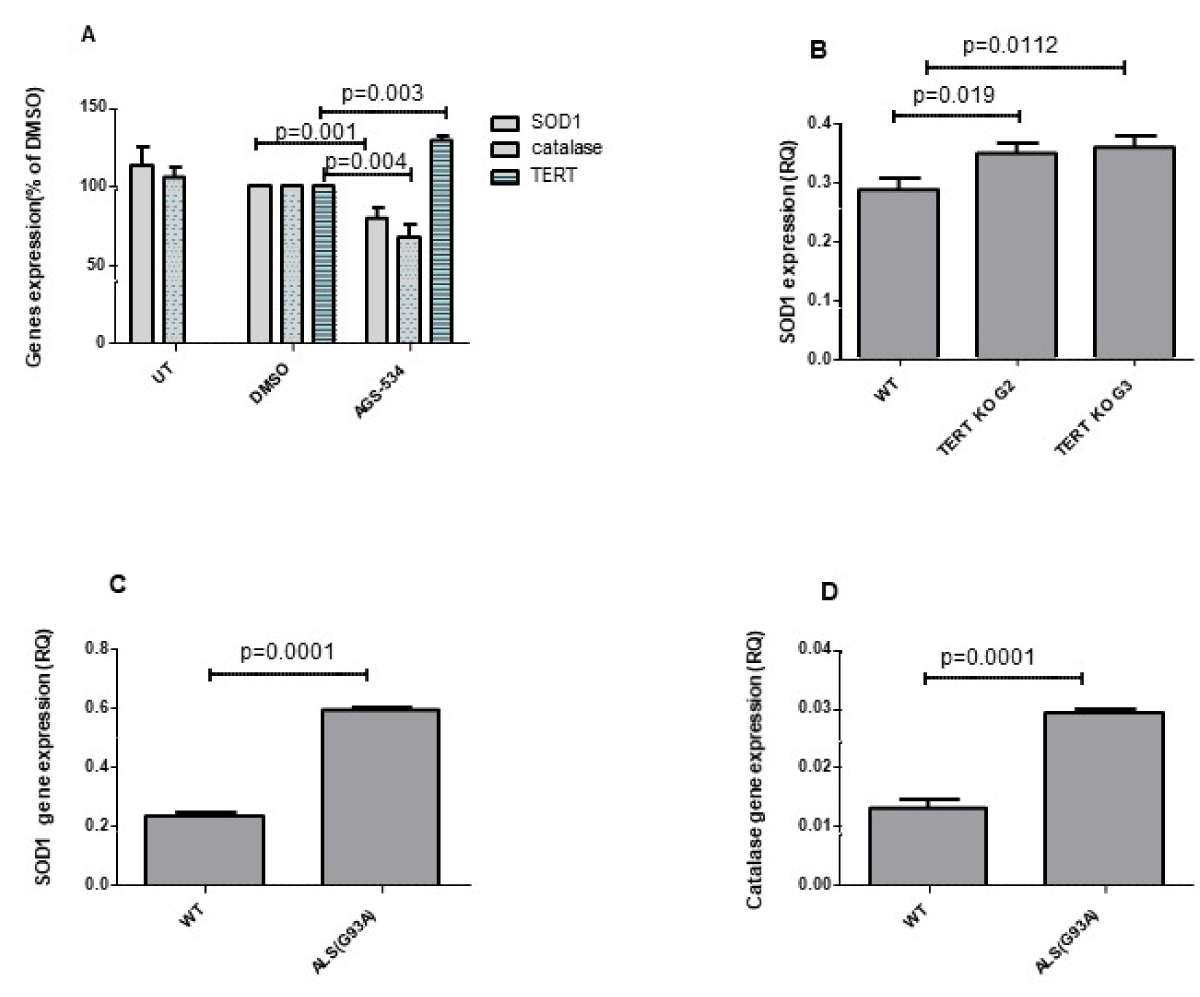
Figure 5: SOD1 and catalase expression increased in the brain of TERT KO mice and decreased in WT brain treated with AGS (A). ICR WT mice were S.C. injected with a single dose of AGS compound (6 mg/Kg) or with vehicle (0.5% DMSO) and sacrificed 12 hrs. later. SOD1, Catalase, and mTERT expression were measured in the brain RNA extracts by Real-time PCR with the appropriate primers. The results are % of vehicle and mean ±SEM of 3 - 5 mice per group (A) p values were calculated by one-way ANOVA test. SOD1 expression in the brains of TERT KO G2 and G3 mice and their WT counterparts was examined using real-time PCR with the appropriate primers. The results are manse of 5 -10 mice per group. p values were calculated by the student T-test (C and D) The brain from hSOD1 Tg mice (ALS) and their WT counterparts were subjected to RNA extraction and the level of the endogenous SOD1, and catalase expression was evaluated by real-time PCR with the appropriate primers. The results are presented as relative quantity and are mean ±SEM of 3 hSOD1 Tg mice and 5 WT mice. p values were calculated by the Student T test.
Several studies illustrated the therapeutic benefits of the telomerase-based approach in ALS and other neurodegenerative diseases particularly the neuroprotective effects of TERT in the brain [19,20-23,39]. This can be attributed to two distinct capabilities of TERT: Its ability to elongate telomeres, thus supporting cell division and cellular life span, which is less relevant to long-lived postmitotic cells as neurons but may affect the disease through neuroinflammation and other glia-related functions; and its protective effect from oxidative stress which was demonstrated in several independent groups at various cell types and animal models including embryonic hippocampal and motor neuron primary culture [14,19,22,36].
These two capabilities are related since it was shown that oxidative stress interferes with telomere maintenance at two levels: it increases the rate of telomeres shortening [40], and it prevents telomerase from counteracting telomere shortening by inducing its export from the nucleus into the mitochondria, where it protects mitochondrial DNA from oxidative damage and inhibits cellular apoptosis [24,41]. Since Motor neurons are non-dividing cells, their telomeres will get shorter mostly due to DNA damage. In addition, ALS is characterized by severe oxidative damage [5]. Therefore, it is reasonable to suggest that the neuroprotective effect of TERT is mostly mediated by reducing oxidative stress rather than maintaining telomeres length. We showed that hTERT confers an anti-apoptotic effect from oxidative stress on human and mouse cells as well [21,36], suggesting that TERT possesses an intrinsic protective mechanism which is not dependent on the external environment. Hence, we investigated the mechanism by which TERT, regardless of mouse or human origin, mediates protection from oxidative stress in a mouse motor neuron-like cell line, NSC-34.
To this end, NSC-34 cells were manipulated by knockdown of mTERT and then knocked-in of hTERT. The downregulation of mTERT resulted in an impaired growth and proliferation capacity, which were not restored by the knocked-in of hTERT. This implies that, although hTERT can interact with the mTR to possess telomerase activity in-vitro, it does not support telomere maintenance in-vivo. These results are in agreement with a previous report [42,43] and highlight the complex regulation of telomerase ribonucleoprotein complex assembly and trafficking to telomeres.
We used an early generation of NSC34mTERT-shRNA cells that did not demonstrate an impaired growth capacity, to examine the cells’ sensitivity to H2O2 insult. The mTERT knocked-down cells had a significantly higher sensitivity to H2O2. hTERT Knocked-in only partially compensated for the loss of mTERT. The results suggest that mTERT can protect mouse motor neurons from oxidative damage and that hTERT cannot fully replace mTERT loss of function.
We were interested in the mechanism of mTERT cellular protection. H2O2 pathology is mediated by ROS, both by itself and by inducing mitochondrial ROS production. Under normal conditions, the ROS levels were similar in the WT and the knocked-down cells. However, in the presence of TERT (in WT and TERT knock-in NSC34 cells), following H2O2 treatment ROS levels were lower compared to their level in mTERT knocked-down cells. This implies that the effect of TERT on ROS reduction is reflected and more prominent under oxidative stress conditions.
Various reports indicate that the presence of TERT in the mitochondria ameliorates mitochondrial function [24,25,42,43]. Therefore, we examined if the increase in cellular ROS levels, following knocking-down of mTERT, is due to mitochondrial dysfunction.
In the absence of H2O2, mTERT knocked down resulted in a significant increase in the MMP is relative to the WT cells. However, under the H2O2-induced stress condition, the MMP was significantly reduced in the mTERT knocked-down cells, while no significant change was observed in the WT cells. Knocking-in hTERT in the mouse motor neurons did not improve the altered MMP caused by the downregulation of mTERT. To confirm these results, we measured the rate of MMP collapse using the protonophore FCCP. The rate of MMP collapse is in direct correlation with the PMF generated in the mitochondrial intermembrane space by the respiratory chain [44]. Compatibly, relative to the WT cells, the mTERT knocked-down cells showed higher PMF, regardless of the presence of hTERT. Higher PMF is generated by an increased activity rate of the respiratory chain, which may lead to an increased ROS production unless the respiratory chain is better coupled to ATP generation [44]. Examining the intracellular ATP levels revealed that downregulation of mTERT reduced ATP levels, which were not compensated by hTERT. Abnormality in any of these processes can be designated as mitochondrial dysfunction [44]. The reduction in ATP levels can explain the increased sensitivity of the NSC-34mTERT-shRNA cells to the insult of H2O2, as many of the cellular repair mechanisms are dependent on energy consumption. In addition, this reduction is compatible with the impaired growth capacity of the NSC-34mTERT-shRNA and NSC34mTERT-shRNA hTERT cells compared to the NSC34WT cells.
The ability of TERT to improve coupling of the MMP and ATP generation, which reduced ROS formation and thus enabled the cells to survive under oxidative stress was previously reported in human fibroblast and endothelial cells [24]. Our results show that mTERT possesses a similar function and it requires species commutability between the TERT and the mitochondria. This notion is supported by a previous study reporting that mouse and human mitochondrial fusion do not survive on selective medium and lose mitochondrial activity due to incompatibilities between the human mtDNA sequence and the nuclear-encoded mouse mitochondrial proteins [45].
Changes in cellular ROS levels can be attributed to either alteration in ROS production or in ROS degradation. We examined the effect of TERT expression on the expression and activity of selected cytosolic antioxidant enzymes (Catalase, SOD1). Relative to the WT cells, down-regulation of mTERT significantly increased the expression of SOD1 and catalase. However, hTERT knocked-in cells prevented the increase in catalase expression and even demonstrated a significant decrease in SOD1 mRNA and protein expression compared to the WT cells. We assume that the increased expression levels of these antioxidant genes represent a deterioration of the cellular redox state.
This is supported by several reports demonstrating an increase in these genes’ expression levels due to H2O2 exposure [46-48].
The changes in enzyme expression levels are not necessarily correlated with changes in its activity. Since an active enzyme is needed for the cellular adaptive response to any environmental challenge, we examined the effect of TERT on the activities of catalase and SOD1 under oxidative and non-oxidative stress conditions. Measuring Catalase activity in the NSC-34 cells revealed that under non-oxidative stress conditions knocking down of mTERT resulted in a significant increase in catalase activity compared to the WT cells or the hTERT knocked-in cells. This shows that TERT is necessary for the cells to cope with the normal ROS levels but in its absence, the cell increases the activity of antioxidant enzymes such as catalase and SOD1.
H2O2 increases cellular Catalase activity as an intrinsic protective mechanism [48,49]. Examining the effect of 20 µM H2O2 on Catalase activity revealed an increase in the mTERT knocked-down, hTERT knocked-in, and WT H2O2 treated cells, compared to the untreated cells. However, the extent of the increase differed between the cells. This may imply that TERT does not regulate the oxidative stress-induced increase in catalase activity.
Examination of the effect of TERT on the activity of another cytosolic oxidative stress enzyme, SOD1, indicates that under normal conditions or oxidative stress conditions, the activity was significantly reduced in mTERT knocked-down cells, but restored when hTERT was knocked-in. This suggests that the activity of SOD1 is regulated by TERT independent of ROS formation. Interestingly, SOD1 gene expression was increased in mTERT-depleted cells while SOD1 activity decreased independently on oxidative stress exposure. This suggests that in the presence of TERT SOD1 undergoes post-translational modifications that reduced its activity. ROS toxicity is partly achieved by the induction of DNA damage. We have previously found that the expression of hTERT in human Mesenchymal Stem cells (hMSC) and of mTERT in mouse Embryonic Stem cells (mESC) significantly reduced the H2O2 induced- DNA γH2AX foci, that mark DNA lesions [21,36].
To confirm the effect of TERT on SOD1 and catalase expression in vivo in mouse brain we used the AGS compounds which we previously showed their ability to increase TERT expression and activity in various regions of the mouse brain [21] and to protect neurons from oxidative stress in a TERT dependent manner [19,21]. Elevation of TERT in the mouse brain indeed decreased the expression of catalase and SOD1. This is compatible with the in vitro data showing that transfection of TERT-depleted neuronal cells with hTERT decreased the expression of these enzymes. Moreover, examination of SOD1 expression in the TERT KO mouse brain revealed an increase in its expression compared to WT mice. These data are also compatible with the in vitro results in motor neurons depleted TERT. The neuroprotective effect of TERT from oxidative stress reduced the amount of ROS and therefore the level of the intrinsic anti-oxidative enzyme is reduced, as can be seen in vitro and in vivo, while depletion of TERT increased ROS level and therefore induced the expression of the intrinsic antioxidant enzymes.
Altogether, our results show that the TERT protective effect is pleiotropic: 1. Reduced ROS generation by improving mitochondrial functionality. 2. Increased ATP levels that enable better defense mechanisms activation. 3. Regulation of the expression of antioxidative enzymes, probably not directly but rather due to the reduction in the amount of ROS.
hTERT only slightly affects catalase activity in mTERT knocked-down cells, suggesting that the effect of TERT on catalase is species-specific. In contrast, SOD1 activity was regulated by both mTERT and hTERT.
The ability of TERT to moderate cellular oxidative stress and to affect SOD1 activity and expression is particularly important for motor neurons considering the distinctive features of these cells. (1) The motor neurons are exceptionally sensitive to SOD1 mutations, which may lead to familial ALS disease [50]. SOD1 depletion cause defects in motor neuron specifically (2) Motor neuron are among the largest cells in the body that deliver messages through long axons, which require a high level of mitochondria and energy. The ability of mTERT to improve mitochondrial functionality and to increase ATP level may be important for the function of these cells and their survival ;(3) We previously demonstrated that motor neurons are significantly more sensitive to oxidative stress compared to other cells such as hMSC or mESC. Comparing the dose of H2O2 required for substantial cytotoxic effect in the NSC-34, to human Mesenchymal stem cells (hMSC) or mouse embryonic stem cells (mESC) revealed that the H2O2 CC50 for NSC-34 was 34 µM H2O2 for 1h, while 40% cell death occurred in hMSC and mESC after treatment of H2O2 in a concentration of 300 µM for 4h or 200 µM for 2h, respectively [21,36].
Few studies demonstrated the benefits of telomerase as a therapeutic approach for neurodegenerative diseases based on its functions in telomere maintenance and oxidative stress. In humans, telomerase expression is significantly reduced in the spinal cord of ALS patients. Indeed, we demonstrated the beneficial effects of increasing telomerase by AGS compounds on the onset and progression of ALS in SOD1 Tg mice and the survival of motor neurons. Here we show for the first time the involvement of TERT in the regulation of SOD1 in motor neuron-like cells and in vivo in the mouse brain which strengthens the notion that increasing TERT may improve the ability of motor neurons to cope with oxidative stress and increase their survival in neurodegenerative disease like ALS.
Author contribution
Conceptualization, A.T., E.E., and E.P.; Performed the experiments A.T., E.E., and S.T.; Supervision, E.P., and E.BY.; Writing—original draft, A.E., E.E., and E.P.; Writing— review & editing, E.P., E.BY and E.E. All authors have read and agreed to the published version of the manuscript.
E.P.- filed patents on AGS compounds.
- Mead RJ, Shan N, Reiser HJ, Marshall F, Shaw PJ. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov. 2023 Mar;22(3):185-212. doi: 10.1038/s41573-022-00612-2. Epub 2022 Dec 21. PMID: 36543887; PMCID: PMC9768794.
- Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1998 Nov;71(5):2041-8. doi: 10.1046/j.1471-4159.1998.71052041.x. PMID: 9798929.
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH Jr, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997 Nov;69(5):2064-74. doi: 10.1046/j.1471-4159.1997.69052064.x. PMID: 9349552.
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006 Oct 5;52(1):39-59. doi: 10.1016/j.neuron.2006.09.018. PMID: 17015226.
- Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010 Mar 1;48(5):629-41. doi: 10.1016/j.freeradbiomed.2009.11.018. Epub 2009 Dec 4. PMID: 19969067.
- Bendotti C, Carrì MT. Lessons from models of SOD1-linked familial ALS. Trends Mol Med. 2004 Aug;10(8):393-400. doi: 10.1016/j.molmed.2004.06.009. PMID: 15310460.
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151-73. doi: 10.1146/annurev.neuro.31.061307.090711. PMID: 18558852.
- Peggion C, Scalcon V, Massimino ML, Nies K, Lopreiato R, Rigobello MP, Bertoli A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants (Basel). 2022 Mar 23;11(4):614. doi: 10.3390/antiox11040614. PMID: 35453299; PMCID: PMC9032988.
- De Felice B, Annunziata A, Fiorentino G, Manfellotto F, D'Alessandro R, Marino R, Borra M, Biffali E. Telomerase expression in amyotrophic lateral sclerosis (ALS) patients. J Hum Genet. 2014 Oct;59(10):555-61. doi: 10.1038/jhg.2014.72. Epub 2014 Aug 21. PMID: 25142509.
- Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR. Protein composition of catalytically active human telomerase from immortal cells. Science. 2007 Mar 30;315(5820):1850-3. doi: 10.1126/science.1138596. PMID: 17395830.
- Hockemeyer D, Collins K. Control of telomerase action at human telomeres. Nat Struct Mol Biol. 2015 Nov;22(11):848-52. doi: 10.1038/nsmb.3083. PMID: 26581518; PMCID: PMC4765361.
- Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, McLaughlin M, Veenstra TD, Nusse R, McCrea PD, Artandi SE. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009 Jul 2;460(7251):66-72. doi: 10.1038/nature08137. PMID: 19571879; PMCID: PMC4349391.
- Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT). Gene. 2012 May 1;498(2):135-46. doi: 10.1016/j.gene.2012.01.095. Epub 2012 Feb 13. PMID: 22381618; PMCID: PMC3312932.
- Eitan E, Tichon A, Daniel G, Priel E. Telomerase expression in adult and old mouse Purkinje neurons. Rejuvenation Res. 2012 Apr;15(2):206-9. doi: 10.1089/rej.2011.1285. PMID: 22533433.
- Tichon A, Gowda BK, Slavin S, Gazit A, Priel E. Telomerase activity and expression in adult human mesenchymal stem cells derived from amyotrophic lateral sclerosis individuals. Cytotherapy. 2009;11(7):837-48. doi: 10.3109/14653240903136979. PMID: 19903097.
- Liu J, Baykal A, Fung KM, Thompson-Lanza JA, Hoque A, Lippman SM, Sahin A. Human telomerase reverse transcriptase mRNA is highly expressed in normal breast tissues and down-regulated in ductal carcinoma in situ. Int J Oncol. 2004 Apr;24(4):879-84. PMID: 15010825.
- Yasumoto S, Kunimura C, Kikuchi K, Tahara H, Ohji H, Yamamoto H, Ide T, Utakoji T. Telomerase activity in normal human epithelial cells. Oncogene. 1996 Jul 18;13(2):433-9. PMID: 8710384.
- Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, Brooks MW, Kaneko S, Murakami S, DeCaprio JA, Weinberg RA, Stewart SA, Hahn WC. Telomerase maintains telomere structure in normal human cells. Cell. 2003 Jul 25;114(2):241-53. doi: 10.1016/s0092-8674(03)00550-6. PMID: 12887925.
- Baruch-Eliyahu N, Rud V, Braiman A, Priel E. Telomerase increasing compound protects hippocampal neurons from amyloid beta toxicity by enhancing the expression of neurotrophins and plasticity related genes. Sci Rep. 2019 Dec 2;9(1):18118. doi: 10.1038/s41598-019-54741-7. PMID: 31792359; PMCID: PMC6889131.
- Ding X, Liu X, Wang F, Wang F, Geng X. Role of Senescence and Neuroprotective Effects of Telomerase in Neurodegenerative Diseases. Rejuvenation Res. 2020 Apr;23(2):150-158. doi: 10.1089/rej.2018.2115. Epub 2019 Jul 18. PMID: 31170886.
- Eitan E, Tichon A, Gazit A, Gitler D, Slavin S, Priel E. Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol Med. 2012 Apr;4(4):313-29. doi: 10.1002/emmm.201200212. Epub 2012 Feb 20. PMID: 22351600; PMCID: PMC3376858.
- Wan T, Weir EJ, Johnson M, Korolchuk VI, Saretzki GC. Increased telomerase improves motor function and alpha-synuclein pathology in a transgenic mouse model of Parkinson's disease associated with enhanced autophagy. Prog Neurobiol. 2021 Apr;199:101953. doi: 10.1016/j.pneurobio.2020.101953. Epub 2020 Nov 11. PMID: 33188884; PMCID: PMC7938226.
- Yu X, Liu MM, Zheng CY, Liu YT, Wang Z, Wang ZY. Telomerase reverse transcriptase and neurodegenerative diseases. Front Immunol. 2023 Mar 29;14:1165632. doi: 10.3389/fimmu.2023.1165632. PMID: 37063844; PMCID: PMC10091515.
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008 Apr 1;121(Pt 7):1046-53. doi: 10.1242/jcs.019372. Epub 2008 Mar 11. PMID: 18334557.
- Haendeler J, Dröse S, Büchner N, Jakob S, Altschmied J, Goy C, Spyridopoulos I, Zeiher AM, Brandt U, Dimmeler S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009 Jun;29(6):929-35. doi: 10.1161/ATVBAHA.109.185546. Epub 2009 Mar 5. PMID: 19265030.
- Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W, Cheung P, Jun S, Artandi MK, Shah N, Kim SK, Artandi SE. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008 Jan;4(1):e10. doi: 10.1371/journal.pgen.0040010. Epub 2007 Dec 13. PMID: 18208333; PMCID: PMC2211538.
- Martens A, Schmid B, Akintola O, Saretzki G. Telomerase Does Not Improve DNA Repair in Mitochondria upon Stress but Increases MnSOD Protein under Serum-Free Conditions. Int J Mol Sci. 2019 Dec 19;21(1):27. doi: 10.3390/ijms21010027. PMID: 31861522; PMCID: PMC6981674.
- Matusica D, Fenech MP, Rogers ML, Rush RA. Characterization and use of the NSC-34 cell line for study of neurotrophin receptor trafficking. J Neurosci Res. 2008 Feb 15;86(3):553-65. doi: 10.1002/jnr.21507. PMID: 17896795.
- Regev L, Ezrielev E, Gershon E, Gil S, Chen A. Genetic approach for intracerebroventricular delivery. Proc Natl Acad Sci U S A. 2010 Mar 2;107(9):4424-9. doi: 10.1073/pnas.0907059107. Epub 2010 Feb 8. PMID: 20142482; PMCID: PMC2840118.
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994 Dec 23;266(5193):2011-5. doi: 10.1126/science.7605428. PMID: 7605428.
- Gething MJ, McCammon K, Sambrook J. Protein folding and intracellular transport: evaluation of conformational changes in nascent exocytotic proteins. Methods Cell Biol. 1989;32:185-206. doi: 10.1016/s0091-679x(08)61171-1. PMID: 2691850.
- Kaufmann SH, Svingen PA. Immunoblot analysis and band depletion assays. Methods Mol Biol. 1999;94:253-68. doi: 10.1385/1-59259-259-7:253. PMID: 12844881.
- Aranda A, Sequedo L, Tolosa L, Quintas G, Burello E, Castell JV, Gombau L. Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay: a quantitative method for oxidative stress assessment of nanoparticle-treated cells. Toxicol In Vitro. 2013 Mar;27(2):954-63. doi: 10.1016/j.tiv.2013.01.016. Epub 2013 Jan 26. PMID: 23357416.
- Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 2010;594:57-72. doi: 10.1007/978-1-60761-411-1_4. PMID: 20072909.
- Joshi DC, Bakowska JC. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J Vis Exp. 2011 May 23;(51):2704. doi: 10.3791/2704. PMID: 21654619; PMCID: PMC3143685.
- Tichon A, Eitan E, Kurkalli BG, Braiman A, Gazit A, Slavin S, Beith-Yannai E, Priel E. Oxidative stress protection by novel telomerase activators in mesenchymal stem cells derived from healthy and diseased individuals. Curr Mol Med. 2013 Jul;13(6):1010-22. doi: 10.2174/1566524011313060013. PMID: 23590815.
- Muir JK, Tynan M, Caldwell R, Ellis EF. Superoxide dismutase improves posttraumatic cortical blood flow in rats. J Neurotrauma. 1995 Apr;12(2):179-88. doi: 10.1089/neu.1995.12.179. PMID: 7629864.
- Tada-Oikawa S, Oikawa S, Kawanishi M, Yamada M, Kawanishi S. Generation of hydrogen peroxide precedes loss of mitochondrial membrane potential during DNA alkylation-induced apoptosis. FEBS Lett. 1999 Jan 8;442(1):65-9. doi: 10.1016/s0014-5793(98)01618-4. PMID: 9923606.
- González-Giraldo Y, Forero DA, Echeverria V, Gonzalez J, Ávila-Rodriguez M, Garcia-Segura LM, Barreto GE. Neuroprotective effects of the catalytic subunit of telomerase: A potential therapeutic target in the central nervous system. Ageing Res Rev. 2016 Jul;28:37-45. doi: 10.1016/j.arr.2016.04.004. Epub 2016 Apr 16. PMID: 27095058.
- Petersen S, Saretzki G, von Zglinicki T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp Cell Res. 1998 Feb 25;239(1):152-60. doi: 10.1006/excr.1997.3893. PMID: 9511733.
- Gordon DM, Santos JH. The emerging role of telomerase reverse transcriptase in mitochondrial DNA metabolism. J Nucleic Acids. 2010 Sep 21;2010:390791. doi: 10.4061/2010/390791. PMID: 20936168; PMCID: PMC2945669.
- Massard C, Zermati Y, Pauleau AL, Larochette N, Métivier D, Sabatier L, Kroemer G, Soria JC. hTERT: a novel endogenous inhibitor of the mitochondrial cell death pathway. Oncogene. 2006 Aug 3;25(33):4505-14. doi: 10.1038/sj.onc.1209487. Epub 2006 Apr 17. Erratum in: Oncogene. 2013 Jan 24;32(4):536. PMID: 16619047.
- Saretzki G. Telomerase, mitochondria and oxidative stress. Exp Gerontol. 2009 Aug;44(8):485-92. doi: 10.1016/j.exger.2009.05.004. Epub 2009 May 18. PMID: 19457450.
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011 Apr 15;435(2):297-312. doi: 10.1042/BJ20110162. Erratum in: Biochem J. 2011 Aug 1;437(3):575. PMID: 21726199; PMCID: PMC3076726.
- Yoon YG, Haug CL, Koob MD. Interspecies mitochondrial fusion between mouse and human mitochondria is rapid and efficient. Mitochondrion. 2007 May;7(3):223-9. doi: 10.1016/j.mito.2006.11.022. Epub 2006 Dec 9. PMID: 17251069; PMCID: PMC2693707.
- Godon C, Lagniel G, Lee J, Buhler JM, Kieffer S, Perrot M, Boucherie H, Toledano MB, Labarre J. The H2O2 stimulon in Saccharomyces cerevisiae. J Biol Chem. 1998 Aug 28;273(35):22480-9. doi: 10.1074/jbc.273.35.22480. PMID: 9712873.
- Lee J, Godon C, Lagniel G, Spector D, Garin J, Labarre J, Toledano MB. Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J Biol Chem. 1999 Jun 4;274(23):16040-6. doi: 10.1074/jbc.274.23.16040. PMID: 10347154.
- Martins D, English AM. Catalase activity is stimulated by H(2)O(2) in rich culture medium and is required for H(2)O(2) resistance and adaptation in yeast. Redox Biol. 2014 Jan 10;2:308-13. doi: 10.1016/j.redox.2013.12.019. PMID: 24563848; PMCID: PMC3926110.
- Ebert R, Ulmer M, Zeck S, Meissner-Weigl J, Schneider D, Stopper H, Schupp N, Kassem M, Jakob F. Selenium supplementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro. Stem Cells. 2006 May;24(5):1226-35. doi: 10.1634/stemcells.2005-0117. Epub 2006 Jan 19. PMID: 16424399.
- Berdyński M, Miszta P, Safranow K, Andersen PM, Morita M, Filipek S, Żekanowski C, Kuźma-Kozakiewicz M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci Rep. 2022 Jan 7;12(1):103. doi: 10.1038/s41598-021-03891-8. PMID: 34996976; PMCID: PMC8742055.